










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMEDISAN
versión On-line ISSN 1029-3019
MEDISAN vol.16 no.12 Santiago de Cuba dic. 2012
CASO CLÍNICO
Nesidioblastosis: hipoglucemia hiperinsulínica persistente en un recién nacido
Nesidioblastosis: persistent hyperinsulinemic hypoglycemia in a newborn
MsC. José Raúl Zaldívar Ochoa,I MsC. Alejandro Rodríguez Carballo,I MsC. Maiyelin Quesada Cortés,II Lic. Marelys Martínez Álvarez,I Dra. Aliniurkis Santiago OconnorI y MsC. Margarita Menéndez RodríguezI
I Hospital Infantil Norte "Dr. Juan de la Cruz Martínez Maceira", Santiago de Cuba, Cuba.
II Policlínico Universitario "Josué País García", Santiago de Cuba, Cuba.
RESUMEN
Se presenta el caso clínico de un recién nacido a término por parto distócico (cesárea), debido a una toxemia gravídica, con antecedente de polihidramnios, que manifestó dificultad respiratoria a los pocos minutos del nacimiento y fue ingresado en el Hospital Infantil Norte "Dr. Juan de la Cruz Martínez Maceira" de Santiago de Cuba. Le fueron detectadas cifras de glucemia muy bajas que, evolutivamente, trajeron aparejadas convulsiones tónico-clónicas generalizadas, cuyas frecuencias no se lograban disminuir. Se le diagnosticó una hipoglucemia hiperinsulínica persistente (nesidioblastosis) y fue trasladado a La Habana para recibir tratamientos clínico y quirúrgico definitivos. Actualmente es atendido por un equipo médico multidisciplinario.
Palabras clave: recién nacido, hiperinsulinismo, hipoglucemia hiperinsulínica persistente, nesidioblastosis, hospitales pediátricos.
ABSTRACT
The case of a term infant by dystocia (cesarean section) due to pregnancy toxemia with history of polyhydramnios is presented, who had respiratory distress a few minutes after birth and he was admitted to "Dr. Juan de la Cruz Martínez Maceira" Northern Children Hospital of Santiago de Cuba. Very low blood glucose levels were detected that progressively caused tonic-clonic seizures, which frequencies could not be reduced. He was diagnosed with persistent hyperinsulinemic hypoglycemia (nesidioblastosis) and was transferred to Havana to receive definitive clinical and surgical treatments. Currently, he is treated by a multidisciplinary medical team.
Key words: newborn, hyperinsulinism, persistent hyperinsulinemic hypoglycemia, nesidioblastosis, children hospitals.
INTRODUCCIÓN
El hiperinsulinismo neonatal constituye la causa más común de hipoglucemia persistente en lactantes. Aunque 95 % de los casos son esporádicos, con una incidencia estimada de 1 por cada 50 000 nacidos vivos y en poblaciones con tasas altas de consanguinidad, puede elevarse a 1 por cada 2 500 nacidos vivos. 1,2
Se refiere que la mayoría de los casos de hiperinsulinismo corresponde a una hiperplasia difusa de células Β, la cual constituye la forma más frecuente del trastorno y afecta a 75 % de los niños.2
De hecho, la hipoglucemia es un riesgo durante los primeros 3 días de vida en, aproximadamente, 80 % de los recién nacidos muy afectados, y puede presentarse en la mayoría de los lactantes antes de los 6 meses de edad. La alteración insulínica puede ser familiar o esporádica y resulta del daño al estímulo de secreción de insulina de las células beta del páncreas. Asimismo, se han notificado mutaciones genéticas en su aparición, además de hiperamonemia --defecto que conduce a la disregulación de dehidrogenasa glutámica--.3-6
Actualmente, gracias al trabajo de muchos investigadores, se han notificado 8 sitios en el organismo asociados al hiperinsulinismo: ABCC8, KCNJ11, HADH1, GCK, GLUD1, SLC16A1, UCP2 y HNF4a. Las mutaciones de estos sitios tienen diferencias importantes en el fenotipo y el modelo de herencia.
Al respecto, los 2 primeros ABCC8 y genes de KCNJ11 se localizan juntos en el brazo corto del cromosoma 11 e involucra a los que presentan alteraciones del canal de potasio en la célula beta, lo cual la conduce a su despolarización con el ingreso de calcio y la consiguiente liberación de insulina. Por otra parte, el canal de potasio más adenosintrifosfato está formado por 2 pares de proteínas denominadas sulfonilureas (Sur-1 y Kir-6.2). Estas 4 subunidades forman un canal de potasio que se cierra en presencia de Atp. Dicha relación de defectos del receptor de sulfonilureas en la membrana de las células beta, con la consecuente alteración del flujo de potasio (ya mencionado) y el incremento de insulina no regulada por los mecanismos habituales de retroalimentación, da como resultado la hipoglucemia persistente. Estas mutaciones son causantes de la forma focal del hiperinsulinismo congénito.7,8
Las mutaciones en el HADH1, con código SCHAD (enzima de oxidación de ácidos grasos), causa la única forma recesiva de hiperinsulinismo. También las mutaciones de GCK, que actúan en la glucoquinasa, causan un hiperinsulinismo que reacciona pobremente al tratamiento médico. Otras formas dominantes de hiperinsulinismo incluyen las mutaciones GLUD1, que actúa en el glutamato de hidrogenasa, lo cual es un paso importante en la estimulación de aminoácidos, la secreción de insulina, y vuelve inactivas las mutaciones UCP2.9
Cabe decir que el diazóxido es una droga que interviene como un agonista en el canal de potasio ATP dependiente y suprime la secreción de insulina. Esta es eficaz en defectos asociados con las mutaciones de GLUD1, UCP2, HNF4a y HADH1; sin embargo, a menudo es ineficaz en las mutaciones del canal de potasio ATP dependiente (ABCC8 y KCNJ11) y no puede controlar el hipoglucemia adecuadamente en GCK o en mutaciones de SLC16A1.9
Precisando de una vez, la hipersecreción de insulina por las células beta del páncreas provoca hipoglucemia persistente sintomática grave, la cual representa un importante problema de salud, puesto que los pacientes requieren soluciones endovenosas con concentraciones elevadas de glucosa (de 25 a 50 %) en infusiones continuas de 10 a 15 mg/kg por minuto y, generalmente, es necesaria la aplicación adicional de hidrocortisona. En ocasiones, el tratamiento convencional no suele ser suficiente y debe optarse por la pancreatectomía subtotal en 90 a 95 % de los afectados.10,11
CASO CLÍNICO
Se describe el caso de un recién nacido a término (37 semanas) por parto distócico (cesárea) debido a una toxemia gravídica, pues la madre era hipertensa y, además, durante el embarazo presentó polihidramnios. No refirió tratamiento médico, ni consanguinidad. El peso al nacer fue de 4 020 gramos (macrosómco), la talla, de 53 cm y el índice de Apgar, de 9/9.
A los pocos minutos de nacido comenzó a presentar dificultad respiratoria, lo que motivó a realizarle varias determinaciones sanguíneas y se hallaron cifras de glucemias muy bajas, a saber: 14,4; 17,8; 24,5 y 34,2 mg/dL; algunas de las cuales trajeron aparejadas convulsiones tónico-clónicas generalizadas, de difícil atención y control, que requirieron cargas iniciales de glucosa endovenosa de 4,8 mg/kg por minuto, aunque evolutivamente continuaron los bajos valores de glucemias.
Ante el cuadro clínico anterior, se decidió incrementar la infusión a 15 mg/kg por minuto, con una concentración de dextrosa a 30 %; además, se incluyeron otros medicamentos como el glucagón, pero no se tuvo una respuesta favorable. Posteriormente, se asoció la hidrocortisona en dosis iniciales de 6 mg/kg diariamente, no obstante, hubo necesidad de aumentar la dosificación hasta 15 mg/kg, porque se mantenían muy bajas las cifras de glucemias --aún unidas a convulsiones--.
Se llegó al consenso médico de que el diagnóstico era: hipoglucemia hiperinsulínica persistente del recién nacido (nesidioblastosis), por los síntomas y signos de la entidad clínica, la respuesta a los diferentes esquemas terapéuticos y los exámenes complementarios realizados. Asimismo, se determinó su traslado inmediato a La Habana para tratarle con diazóxido u octreótido, y aplicarle tratamiento quirúrgico definitivo.
EXÁMENES COMPLEMENTARIOS
Para tratar de identificar el origen de la hipoglucemia persistente, se efectuaron pruebas de laboratorio y otros estudios:
- Determinación de glucemias a través del perfil glucémico: menor de 40 mg/dL
- Examen de orina: cetonuria negativa
- Determinación de hormonas tiroideas: valores normales
- Determinación de insulina en sangre: 48 μU/mL
- Ecografía abdominal: normal
- Tomografía axial computarizada de páncreas: normal
- Índice de glucosa/insulina: glucemia 30 mg/dL ÷ insulina 48 μU/mL= 0,62
Se realizaron además otros estudios: biometría hemática, electrólitos séricos, calcio, fósforo y magnesio, así como las pruebas de función hepática y las metabólicas en orina.
Tras la confirmación del diagnóstico, se inició la medicación con diazóxido en dosis de hasta 20 mg/kg cada día, pero no se lograron disminuir las frecuencias de las hipoglucemias ni de las convulsiones. Presentó varias complicaciones, tales como: neumonías, infecciones micóticas, hipertensión arterial y efecto Cushing secundario a la aplicación del esteroide y diazóxido, por lo cual recibió múltiples tratamientos antimicrobianos, antimicóticos y antihipertensivo. Al no reaccionar favorablemente a la terapia medicamentosa, se decidió operarle (pancreatectomía subtotal) a los 2 meses de edad. Tuvo una estadía hospitalaria de 6 meses.
Actualmente, con 10 meses de edad, tiene un seguimiento clínico por un equipo médico multidisciplinario y se le administra una dieta especial. Desde el punto de vista clínico mantiene, en ocasiones, hipoglucemias sintomáticas e insuficiencia exocrina --tratada con enzimas pancreáticas-- y neurológicamente está muy afectado.
• Antecedentes patológicos personales: No presentaba.
• Antecedentes patológicos familiares: No se refirieron hipoglucemias en familiares cercanos.
EXAMEN FÍSICO
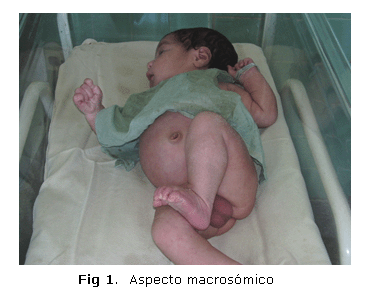
Antes de ser intervenido, se obtuvo una evaluación nutricional mayor de 97 percentil, o sea, su tamaño era macrosómico (figura 1).
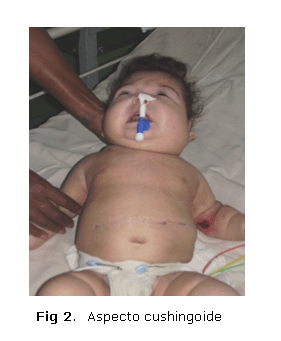
Luego del tratamiento quirúrgico, se observaban características cushingoides (figura 2). Presentaba, además, la herida quirúrgica abdominal y escara en la parte interna del codo izquierdo.
COMENTARIOS
Recientemente se ha propuesto la nifedipina en dosis inicial de 0,3 mg/kg al día hasta llegar a 0,8 mg/kg. Dicho esquema terapéutico se basa en el ingreso del calcio, el cual produce secreción de insulina, pues en los pacientes con nesidioblastosis el cierre del canal de potasio despolariza la célula y permite el ingreso del calcio, con la consiguiente secreción de insulina. Al respecto, los antagonistas del calcio podrían facilitar el tratamiento incluso en la etapa neonatal.
Con respecto a la evolución de estos pacientes, los riesgos a largo plazo están dados en la persistencia de hiperinsulinismo, que conduce a una segunda o tercera reintervención, y más tardíamente, la diabetes mellitus y los trastornos de absorción intestinal. En general, quedan muy afectados neurológicamente y poseen una supervivencia muy corta.
REFERENCIAS BIBLIOGRÁFICAS
1. Lee PJ, Leonard JV. Hypoglycemia. En: Brook GD, Clayton P, Brown R. Brook´s Clinical Pediatric Endocrinology. 3 ed. London: Blackwell Science; 1995. V 35. p. 677-93.
2. Dacou-Voutetakis C, Psychou F, Maniati-Christidis M. Persistent hyperinsulinemic hypoglycemia of infancy: long-term results. J Pediatr Endocrinol Metab. 1998; 11(Suppl 1): 131-41.
3. Sinha S, Kwok-Chun Chan K, Sadeghi-Nejad A. Hyperinsulinism [citado 30 Dic 2011]. Disponible en: http://www.emedicine.com/PED/topic1075.htm
4. Aynsley-Green A, Hussain K, Hall J, Saudubray JM, Nihoul-Fékété C, De Lonlay-Debeney P, et al. Practical management of hyperinsulinism in infancy. Arch Dis Child Fetal Neonatal Ed. 2000; 82(2): 98-107.
5. James C, Kapoor RR, Ismail D, Hussain K. The genetic basis of congenital hyperinsulinism. J Med Genet. 2009; 46(5): 289-99.
6. Arnoux JB, de Lonlay P, Ribeiro MJ, Hussain K, Blankenstein O, Mohnike K, et al. Congenital hyperinsulinism. Early Hum Dev. 2010; 86(5): 287-94.
7. Magge SN, Shyng SL, MacMullen C, Steinkrauss L, Ganguly A, Katz LE, et al. Familial leucine-sensitive hypoglycemia of infancy due to a dominant mutation of the beta-cell sulfonylurea receptor. J Clin Endocrinol Metab. 2004; 89(9): 4450-6.
8. Flanagan SE, Kapoor RR, Banerjee I, Hall C, Smith W, Hussain K, et al. Dominantly acting ABCC8 mutations in patients with medically unresponsive hyperinsulinaemic hypoglycaemia. Clin Genet. 2011; 79(6): 582-7.
9. Kapoor RR, James C, Flanagan SE, Ellard S, Eaton S, Hussain K. 3-Hydroxyacyl-coenzyme. A dehydrogenase deficiency and hyperinsulinemic hypoglycemia: characterization of a novel mutation and severe dietary protein sensitivity. J Clin Endocrinol Metab. 2009; 94(7): 2221-5.
10. Lee PJ, Leonard JV. Hypoglycaemia. En: Brook GD. Clinical pediatric endocrinology. 3 ed. London: Blackwell Science; 1995. p. 677-93.
11. Anzoátegui Espínola RE, Dorantes Álvarez LM, García Morales LM, Bracho Blanchet EJ, Garibay Nieto GN, Sadowinski Pine S. Hipoglucemia hiperinsulinémica persistente de la infancia: revisión de casos en un período de 10 años. Bol Med Hosp Infant Mex. 2000; 57(7): 383-9.
Recibido: 10 de abril de 2012.
Aprobado: 24 de junio de 2012.
José Raúl Zaldívar Ochoa. Hospital Infantil Norte "Dr. Juan de la Cruz Martínez Maceira", calle 8, entre 9 y 11, reparto Fomento, Santiago de Cuba, Cuba. Correo electrónico:jzaldivar@medired.scu.sld.cu
