










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMEDISAN
versión On-line ISSN 1029-3019
MEDISAN vol.22 no.1 Santiago de Cuba ene. 2018
CASO CLÍNICO
Telangiectasia hemorrágica hereditaria en una gestante
Hereditary hemorrhagic telangiectasia in a pregnant woman
Dra. Mabel González Escudero, I Dra. Marilyn Sosa Estébanez II y Dr. Noel David Pérez Acosta II
I Hospital General Docente Intermunicipal "Mártires del 9 de Abril", Sagua la Grande, Villa Clara, Cuba.
II Policlínico Docente "Mario A Pérez", Sagua la Grande, Villa Clara, Cuba.
RESUMEN
Se presenta el caso clínico de una embarazada de 16 años de edad, quien acudió a las consultas de dermatología y genética por presentar lesiones cutáneas. Luego de realizar los estudios pertinentes se consideró el diagnóstico de telangiectasia hemorrágica hereditaria. Se remitió al Hospital "Mártires del 9 de Abril" de Sagua La Grande con sangrado vaginal, rectal y de labios, que fue controlado. Se realizaron interconsultas con otros especialistas para garantizar la atención multidisciplinaria durante el embarazo y el parto. La paciente evolucionó favorablemente hasta tener a su bebé a través de un parto eutócico.
Palabras clave: gestante, hemorragia hereditaria, síndrome de RenduOsler-Weber, malformación arteriovenosa, atención secundaria de salud.
ABSTRACT
The case report of a 16 years pregnant woman is presented who went to the dermatology and genetics services due to cutaneous lesions. After carrying out the pertinent studies, the diagnosis of hereditary hemorrhagic telangiectasia was considered. She was referred to "Mártires del 9 de Abril" Hospital in Sagua La Grande with vaginal, rectal and labium bleeding that was controlled. Some consultations with other specialists were carried out to guarantee the multidisciplinary care during pregnancy and childbirth. The patient had a favorable clinical course until having her baby through an eutocic childbirth.
Key words: pregnant woman, hereditary hemorrhages, Rendu-Osler-Weber syndrome, arteriovenous malformation, secondary health care.
INTRODUCCIÓN
EL síndrome de Rendu-Osler-Weber (SROW) o telangiectasia hemorrágica hereditaria (THH) es un trastorno displásico fibrovascular con manifestaciones en múltiples órganos, que frecuentemente provoca sangrado y se caracteriza por el desarrollo de telangiectasias mucocutáneas y malformaciones arteriovenosas (MAV) viscerales. Es un trastorno genético raro, de carácter autosómico dominante, penetrancia y expresión variable, no relacionado con el sexo, pero afecta a todas las etnias.1-3
Fue descrito por primera vez en 1876 por John Wickham Legg y, posteriormente, por Henri Jules Rendu y Frederick Parkes Weber, en 1896. Luego, en 1901, William Osler describió el caso de 3 pacientes, quienes presentaban una forma familiar rara de hemorragias nasales recurrentes (epistaxis), asociada a la dilatación de pequeños vasos sanguíneos, que daba lugar a manchas color púrpura con aspecto de araña en piel y mucosas (telangiectasias).4
Se han identificado, al menos, 5 mutaciones en genes que pueden causar THH; estos genes codifican proteínas que modulan la actividad del factor de crecimiento transformante beta (TGF)-β superfamilia de señalización en las células endoteliales vasculares que regulan la proliferación celular, diferenciación, migración y formación de matriz extracelular. Según la afectación se reconocen 5 subtipos diferentes, de los cuales las 2 mutaciones más importantes por su frecuencia (mayor que 80 %) son: en el cromosoma 9 (ENG, gen que produce la endoglina); herencia medelian en el hombre (OMIM, por sus siglas en inglés) No. 187300, relacionado con THH 1 y el cromosoma 12 (proteína: receptor de activina-cinasa tipo 1 ACVRL, por sus siglas en inglés), ALK-1, OMIM No. 600376 relacionado con la THH 2. Una pequeña proporción de los pacientes tienen THH debido a una mutación en el gen MADH4 (producto de proteína: Smad4, OMIM No.175050) como parte de un síndrome de superposición de poliposis juvenil-THH.2
La endoglina participa en procesos tales como activación monocito-macrófago, también en la preeclampsia y en mecanismos vinculados con el cáncer.5
En el 2000, un grupo de expertos se reunió en la Isla de Curazao, donde establecieron una serie de criterios que sirvieron de base para llevar a cabo el diagnóstico clínico de esta enfermedad de forma consensuada. De este modo, los "Criterios de Curazao" se usan actualmente para llevar a cabo el diagnóstico clínico de THH, a saber:
- Epistaxis: espontáneas y recurrentes
- Telangiectasias: múltiples y en sitios característicos (labios, cavidad bucal, dedos, nariz)
- Lesiones viscerales: telangiectasias gastrointestinales, malformaciones arteriovenosas pulmonares, hepáticas o cerebrales
- Historia familiar: pariente de primer grado con THH diagnosticado según los criterios expuestos
Desde el punto de vista clínico, el diagnóstico se considera: definitivo, si presenta 3 o más criterios; posible o sospechoso, 2 de estos criterios; incierto, menos de 2.6
Las telangiectasias mucocutáneas suelen aparecer entre 5 y 20 años después de las epistaxis y aumentan en número con la edad. Los sitios más comunes de localización mucosa son: nariz, vías urinarias, tracto gastrointestinal, laringe, vejiga y vagina.
Por otra parte, las telangiectasias en la piel son superficiales, puntiformes, purpúreas, de aspecto de araña, de 1 a 4 mm de diámetro, que palidecen con la presión y se localizan de forma diseminada en cualquier parte del tegumento cutáneo. Estas lesiones aparecen durante la niñez, aumentan de tamaño en el transcurso de la adolescencia y pueden adquirir forma aracneiforme, así como generar complicación por su tendencia hemorrágica.4
CASO CLÍNICO
Se presenta el caso clínico de una paciente blanca, de 16 años de edad, con 2 gestaciones y un aborto, quien asistió a interconsulta de dermatología por interés científico de la especialista en Genética Clínica, debido a las lesiones cutáneas que presentaba cuando fue examinada a las 13 semanas del embarazo.
Al realizarle el examen cutáneo mucoso llamó la atención:
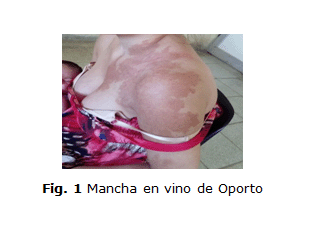
- Mancha en vino de Oporto extensa que ocupaba parte de la cara anterior del tronco, el cuello y todo el miembro superior izquierdo (figura 1).
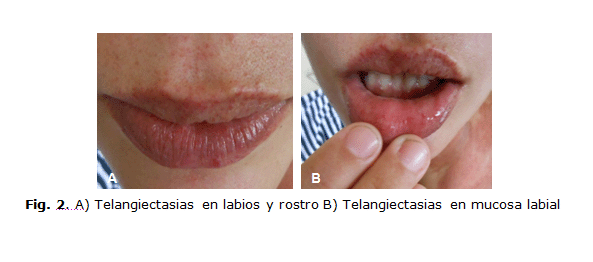
- Lesiones superficiales, puntiformes, purpúreas, de 1 a 4 mm de diámetro que palidecían con la presión y se localizaban en labios, lengua, pulpejos de los dedos, lecho ungueal, dorso de manos y lóbulos de las orejas (figura 2).
Al ser interrogada refirió presentar sangrado nasal frecuente en la infancia, trastornos del ritmo cardiaco con seguimiento por la especialidad de cardiología, pero desconocía si existían otros familiares con igual sintomatología, excepto la figura paterna que tenía lesiones similares en la piel.
En la historia clínica hospitalaria se recogieron los ingresos por forúnculos y ántrax a los 13 y 15 años de edad. Asistió a las interconsultas de Pediatría, Ginecobstetricia, Genética, Cardiología y Hematología -- con la impresión diagnóstica de telangiectasia hemorrágica hereditaria -- para garantizar la atención multidisciplinaria del embarazo y el parto.
Ingresó en el Hogar Materno del municipio hasta las 22,6 semanas donde evolucionó favorablemente. Luego fue remitida al Hospital "Mártires del 9 de Abril" de Sagua La Grande, por presentar sangrado vaginal, rectal y de labios, de poca cuantía, que fue controlado. Recibió atención por el especialista en Gastroenterología, quien evaluó el caso, y se mantuvo en la institución antes mencionada hasta las 37 semanas; clínica y ultrasonográficamente se planteó que el embarazo seguía un curso normal y la THH se mantenía controlada. Se decidió remitirla al Hospital Ginecobstétrico Provincial de la Ciudad de Santa Clara para realizarle el parto transpelviano con la intervención del especialista en hematología.
Durante el trabajo de parto se administró ácido tranexámico por vía endovenosa. Se produjo el parto eutócico a las 40,6 semanas. Nació un feto hembra, de 3700 gramos, con circunferencia cefálica (CC) de 33 cm, talla de 56 cm y test de Apgar: 8-9. Tanto la madre como el bebé evolucionaron favorablemente.
COMENTARIOS
La telangiectasia hemorrágica hereditaria es una displasia vascular de herencia autosómica dominante, caracterizada por telangiectasias cutaneomucosas y malformaciones arteriovenosas en distintos órganos, entre los cuales figuran: cerebro, pulmón, hígado y tubo digestivo.
Comúnmente se presenta epistaxis espontánea y recurrente, anemia ferropénica, así como telangiectasias en cavidad bucal, rostro y miembros superiores. Las familias afectadas presentan una importante morbilidad y mortalidad, así como gran deterioro en su calidad de vida.5
Los estudios complementarios buscan evaluar las malformaciones vasculares más frecuentes. Así, algunos autores4,7 sugieren que deberían incluirse los siguientes exámenes:
• Ecocardiografía y tomografía computarizada con estudio vascular de tórax para analizar malformaciones arteriovenosas pulmonares.
• Resonancia magnética nuclear cerebral para estudiar malformaciones arteriovenosas cerebrales.
• Ecotomografía Doppler de hígado y tomografía computarizada abdominal para estudio de malformaciones arteriovenosas hepáticas. El estudio endoscópico de telangiectasias gastrointestinales podría diferir en individuos asintomáticos y sin anemia.
• Finalmente, el estudio de malformaciones en otros sistemas dependerá de la sospecha clínica.7
• Otros autores señalan que la tomografía de alta resolución combinada con reconstrucción 3D y angiografía digital, es el método diagnóstico de elección por su alta sensibilidad y especificidad.8
El conocimiento de las mutaciones genéticas involucradas en la THH brinda la posibilidad de incorporar técnicas de diagnóstico molecular a la práctica clínica.
Estudios con técnicas de secuenciación directa como la cromatografía líquida de alta precisión han demostrado que sería posible detectar mutaciones características en 70- 90 % de los individuos con la citada enfermedad.7
La mayoría de las embarazadas que presentan THH evolucionan normalmente, aunque existen riesgos de complicaciones potencialmente mortales para la madre y el feto. Dichos embarazos deben ser considerados de alto riesgo y hay que orientar a las pacientes sobre las complicaciones. La generalidad de los casos transcurre sin incidentes, aunque la tasa de mortalidad es significativa, debido a hemorragias de malformaciones arteriovenosas pulmonares, ictus e infartos del miocardio. La supervivencia puede ser mejorada por el reconocimiento previo del estado de la enfermedad y la presencia de malformaciones arteriovenosas pulmonares.
El feto puede presentar complicaciones vasculares, shunt vascular, hemorragias intraútero (pulmonares o cerebrales), hidropesía y pueden aumentar los decesos antes del parto.
Clásicamente se recomendaba el control preparto de las malformaciones arteriovenosas (PMAV), donde estaría justificado el tratamiento de estas durante la etapa preconcepcional o prenatal. El embarazo ha sido reconocido como un factor que favorece el crecimiento y, por ende, la ruptura de las PMAV. En principio, esto se atribuye a los cambios circulatorios que acontecen durante dicha etapa; sin embargo, no existe evidencia suficiente para avalar el tratamiento de las PMAV durante el embarazo. Se recomienda dar seguimiento a las gestantes en las unidades de alto riesgo; ofrecer una educación adecuada y pautas de alarma, así como un parto o cesárea controlados. Idealmente, las PMAV deben ser tratadas en la etapa preconcepcional. De hecho, se descartará la presencia de MAV en columna lumbar antes de cualquier anestesia raquídea o peridural.3
Los autores de este artículo refieren que existe un gran número de especialistas en dermatología, los cuales están muy poco familiarizados con las malformaciones arteriovenosas. Ello se debe, en parte, a la poca prevalencia de dichas lesiones y al hecho de que generalmente suelen ser tratadas por médicos de otras especialidades, en particular, radiólogos intervencionistas de adultos y niños, médicos maxilofaciales y cirujanos plásticos. Las MAV deben atenderse desde una aproximación multidisciplinaria, siendo el objetivo principal de los dermatólogos realizar un correcto diagnóstico para evitar, de este modo, tratamientos innecesarios. En la última década se ha incrementado notoriamente el conocimiento sobre su fisiopatología, lo cual ha permitido el desarrollo de nuevas estrategias terapéuticas.5
Para prevenir las hemorragias se utilizan, entre otros componentes: hierro, ácido tranexámico, fibrinolítico de acción prolongada (2 a 4 gramos diarios), hormonoterapia (estrógenos con progestágenos o sin ellos) o embolización endovascular con partículas, que es el método prescrito en los casos de procesos hemorrágicos profusos y recurrentes.
Se han descrito moduladores de receptores de estrógenos, tales como tamoxifeno y más recientemente raloxifen, los cuales han mostrado resultados favorables. También, se ha ensayado con Bevacizumab, anticuerpo monoclonal recombinante humanizado con terapia de fotocoagulación láser, administrado por inyección submucosa, tópica y vía endovenosa, con resultados variables.
Otros tratamientos farmacológicos incluyen la talidomida, por su efecto antiangiogénico e inductor de la maduración vascular. Se señalan además, aquellos que actúan a nivel de la angiogénesis, como son los betaadrenérgicos y el uso tópico de timolol oftálmico al 0,5 %.7 Se citan también, la cirugía neurovascular, radiocirugía estereotáctica o multimodal.1 El trasplante hepático es hasta el momento el único tratamiento curativo de la afección hepática. 5
La enfermedad de Rendu-Osler-Weber o THH es poco frecuente, con un amplio espectro clínico y un riesgo importante de presentar complicaciones, en particular durante el embarazo y el parto. La atención de esta incluye, la búsqueda activa de malformaciones arteriovenosas y el dermatólogo puede ser, como en el caso presentado, quien estudie y atienda de manera oportuna al paciente.
REFERENCIAS BIBLIOGRÁFICAS
1. Vilela P, Mazza A, Chaumeil P, Miller A, Benavides O. Absceso cerebral en paciente con síndrome de Rendu-Osler-Weber. Revista de la Facultad de Ciencias Médicas. 2016 [citado 5 Abr 2017]; 6(4). Disponible en: http://revista.med.unlp.edu.ar/archivos/201609/absceso-cerebral-en-paciente-con-s%C3%ADndrome-de-rendu-osler-weber.pdf
2. Alonso Gómez M, Fernando Ruiz O, Otero W. Telangiectasia hemorrágica hereditaria. Reporte de caso. Asociaciones Colombianas de Gastroenterología, Endoscopia digestiva, Coloproctología y Hepatología. 2015 [citado 5 Abr 2017]. Disponible en: http://www.scielo.org.co/pdf/rcg/v30n4/v30n4a11.pdf
3. Oramas D, Fuentes H, Arévalo K, Arias L, Ávila C. Trascendencia Perinatal del síndrome de Osler-Weber-Rendu. Rev Latin Perinat. 2012; 15(3):155-8.
4. Bustamante F, Tenreiro Picón O, Tenreiro A, Bustamante E. Síndrome de Rendu-Osler-Weber: presentación de un caso clínico. Avan Biomed. 2016 [citado 5 Abr 2017]; 5(2). Disponible en: http://www.redalyc.org/pdf/3313/33134741700 9.pdf
5. Serra MM. Telangiectasia hemorrágica hereditaria (Síndrome de Rendu-Osler-Weber). Rev Hosp Ital B Aires. 2014 [citado 5 Abr 2017]; 34(2). Disponible en https://www.hospitalitaliano.org.ar/multimedia/archivos/noticias_attachs/47/documentos/17660_41 -50-Revision-Serra.pdf
6. Molgó M, Salomone C, Musalem A, Zuleta A. Telangiectasia hemorrágica hereditaria (enfermedad de Rendu-Osler-Weber): a propósito de un caso. Dermatol Pediatr Lat. 2004 [citado 5 Abr 2017]; 2(2). Disponible en: http://sisbib.unmsm.edu. pe/BVRevistas/dpl/v02n02/PDF/a05.pdf
7. Alvo A, Alzérreca E. Telangiectasia hemorrágica hereditaria: Aspectos otorrinolaringologías. Rev. Otorrinolaringol Cir Cabeza Cuello. 2012 [citado 5 Abr 2017]; 72(3). Disponible en: http://www.scielo.cl/scielo.php? script=sci_arttext &pid=S0718-48162012000300014
8. Duque Estrada L, Castro Gutiérrez N, Larquin Comet J, Junco Bonet MD, Betancourt Reyes G. Enfermedad de Rendu Osler Weber: presentación de un caso. Rev Arch Med Camagüey. 2016 [citado 5 Abr 2017]; 20(6). Disponible en: http://scielo.sld.cu/pdf/amc/v20n6/amc130616.pdf
Recibido: 24 de abril de 2017.
Aprobado: 22 de noviembre de 2017.
Mabel González Escudero. Hospital General Docente Intermunicipal "Mártires del 9 de Abril", Carretera Circuito Norte a Quemado de Güines km 2 1/2. Villa Clara, Sagua la Grande, Cuba. Correo electrónico:noelpa@infomed.sld.cu


{kind=link}
{kind=link}