










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkCorreo Científico Médico
versión On-line ISSN 1560-4381
CCM vol.17 supl.1 Holguín 2013
ARTÍCULO DE REVISIÓN
Actualización en enfermedad de Huntington
Update on Huntington Disease
Jorge Michel Rodríguez Pupo 1, Yuna Viviana Díaz Rojas 2, Yesenia Rojas Rodríguez 3, Yordanis Rodríguez Batista 4, Enriqueta Núñez Arias 5
1. Especialista de Primer Grado en Neurología. Hospital General Universitario Vladimir Ilich Lenin. Holguín. Cuba.
2. Licenciada en Gestión de la Información en Salud. Hospital General Universitario Vladimir Ilich Lenin. Holguín. Cuba.
3. Estudiante de la carrera de Medicina. Universidad de Ciencias Médicas. Holguín. Cuba.
4. Especialista de Primer Grado en Medicina General Integral. Policlínica Comunitaria Rubén Batista Rubio. Cacocum. Holguín. Cuba.
5. Especialista de Primer Grado en Neurofisiología Clínica. Hospital Pediátrico Octavio de la Concepción y de la Pedraja. Holguín. Cuba.
RESUMEN
La enfermedad de Huntington es un trastorno neurodegenerativo trasmitido con rasgo autosómico dominante. La pérdida neuronal selectiva en el estriado produce corea y deterioro cognitivo. Se trata de una enfermedad progresiva que comienza en la mitad de la vida adulta, evoluciona de manera crónica durante muchos años y para la que no existe en la actualidad un tratamiento curativo. Se realizó una revisión general del estado actual de conocimiento de la enfermedad de Huntington, se incluyeron aspectos etiopatogénicos, clínicos, de metodología diagnóstica y opciones terapéuticas.
Palabras clave: enfermedad de Huntington, autosómico dominante, corea, crónica, progresiva.
ABSTRACT
Huntington Disease is a neurodegenerative disorder transmitted as an autosomal dominant trait. Selective neuronal loss in the striatum leads to chorea and cognitive impairment. It is a progressive disease with onset in midlife, which chronically evolves over many years and for which no curative treatment is available today. A review on the current knowledge of Huntington Disease was carried out. Etiopathogenic, clinical, diagnostic methods and treatment options were included.
Keywords: Huntington Disease, autosomal dominant, chorea, chronic, progressive.
INTRODUCCIÓN
La palabra corea proviene del griego ¨κορεια¨ (choreia) que significa danza. Este término fue utilizado por primera vez por el médico alquimista Paracelso (1493- 1541) para describir la Corea o ¨baile de San Vito¨ (Chorea Sancti Viti), la cual probablemente se trató de una forma epidémica de corea histérica, que ocurrió en un contexto de fervor religioso 1.
La corea es un trastorno involuntario del movimiento, tipo hipercinético, caracterizado por movimientos espontáneos, sin propósito, excesivos, abruptos, arrítmicos, no sostenidos, irregulares en tiempo, distribuidos en forma aleatoria, con cambios en velocidad y dirección, que migran de una parte del cuerpo a otra, dando la apariencia de danza. En algunos casos los movimientos pueden ser rápidos y abruptos como en la corea de Sydenham, en otros pueden ser más lentos y fluctuantes, ejemplo de ello, es la enfermedad de Huntington, prototipo clásico de las coreas 2.
Enfermedad de Huntington
La enfermedad de Huntington (EH), conocida también como el “mal de San Vito” fue reconocida en 1872 por el médico norteamericano George Summer Huntington (fig. 1), quien hiciera la primera descripción clínica completa y clara de una enfermedad familiar, cuyos pacientes había estudiado junto a su abuelo y su padre en Long Island, Nueva York. El seguimiento familiar de los afectados condujo posteriormente hasta dos hermanos, que en 1630 partieron con sus familias desde Essex (Inglaterra) hacia Boston (EUA). En los tres siglos siguientes, unos 1000 descendientes padecieron la enfermedad; muchos de ellos fueron acusados de brujería, al ser interpretados sus movimientos anormales como «burla a Jesucristo en la cruz» 3.

La EH se define como un trastorno neurodegenerativo progresivo de transmisión autosómica dominante 4. Clínicamente se caracteriza por la combinación de corea y otros movimientos anormales, deterioro cognitivo progresivo, y síntomas psiquiátricos y conductuales 5.
DESARROLLO
Epidemiología
La enfermedad está distribuida en todo el mundo en igual proporción entre hombres y mujeres. La prevalencia se considera entre 5 y 10 casos por 100.000 habitantes, algo menor en países del este asiático y en la población de raza negra. La incidencia anual varía entre 1 y 4 casos por millón de habitantes. Los estudios genealógicos permiten situar el origen de la enfermedad en el oeste de Europa (Francia, Alemania y Holanda), con posterior dispersión hacia América, Inglaterra, Sudáfrica y Australia. Las mayores tasas de prevalencia se dan en la región del lago de Maracaibo de Venezuela, en la isla de Tasmania (Sur de Australia) y en Moray Firth de Escocia 6.
Etiología
La EH se trata de una de las 10 enfermedades hereditarias autosómicas dominantes, con excepción de la atrofia bulboespinal de Kennedy (herencia recesiva ligada al cromosoma X), producidas por expansión excesiva de tripletes CAG (citosina-adenina-guanina) en sus respectivas proteínas (enfermedades poliglutamínicas o poliQ) 7.
Enfermedad Proteína
Enfermedad de Huntington Huntingtina
Enfermedad de Huntington like 2 (HDL 2) Junctofilina 3
Ataxia espinocerebelosa tipo 1(SCA1) Ataxina 1
Ataxia espinocerebelosa tipo 2(SCA2) Ataxina 2
Ataxia espinocerebelosa tipo 3(SCA3) Ataxina 3
Ataxia espinocerebelosa tipo 6(SCA6) Ataxina 6
Ataxia espinocerebelosa tipo 7(SCA7) Ataxina 7
Atrofia dentatorrubropalidoluysiana (DRPLA) Atrofina 1
Ataxia espinocerebelosa tipo 17(SCA17) Proteína ligadora de TATA
Atrofia bulboespinal de Kennedy Receptor de andrógenos
Una amplia colaboración científica pudo identificar (1993) 8 una mutación expansiva (CAG) en el primer exón del gen IT15 9, situado en el cromosoma 4p16.3. Este gen se expande 210 kb y codifica para la huntingtina, una proteína de 348 kDa, de expresión ubicua en el núcleo o citoplasma de las células de diversos tejidos, incluidas las neuronas. La proteína mutante forma agregados nucleares, pero los mecanismos del proceso de neurodegeneración todavía hoy se desconocen 7,10.
El número de copias de este triplete en un individuo normal es menor de 35. Cuando hay 40 o más repeticiones, se produce la EH. Si las repeticiones están entre 36 y 39 la penetrancia de la enfermedad es incompleta 11-13. Las expansiones entre 40 y 50 repeticiones de CAG son vistas con frecuencia en personas que presentan síntomas entre los 30 y 50 años. La EH juvenil (EHJ) se asocia con casos que sobrepasan las 70 repeticiones.
El laboratory Committee by the Huntington Disease Working Group, en Bethesda, Maryland, propuso la siguiente clasificación 14:
1. Alelos normales: alelos con ≤26 repeticiones CAG, no son patológicos y segregan como repeticiones polimórficas estables en >99% de las meiosis. Los alelos más comunes son los de 17 y 19 repeticiones CAG.
2. Alelos normal-mutados: alelos con 27 a 35 repeticiones CAG, intervalo referido como rango de inestabilidad meiótica o alelos intermedios. No producen el fenotipo de la EH pero pueden ser meióticamente inestables en las células germinales masculinas.
3. Alelos HD con penetrancia reducida: alelos con 36 a 39 repeticiones, meióticamente inestables y que pueden producir la EH.
4. Alelos HD con penetrancia completa: tienen más de 40 repeticiones CAG y producen el fenotipo de la EH.
5. Mosaicismo: se debe a la inestabilidad mitótica y meiótica, y se ha descrito en cerebro y células germinales masculinas, además, parece ser más pronunciado en los casos de inicio juvenil asociados con grandes expansiones 15.
El conocimiento actual de la EH aún no permite predecir con exactitud el momento en que los síntomas van a aparecer, sin embargo, la detección de los portadores de la enfermedad en fases presintomáticas es posible gracias a las técnicas de biología molecular. Aunque existen ciertos problemas legales y éticos en el uso del consejo genético, en estos casos se debe tener en cuenta que permite a los padres calcular la probabilidad de tener un hijo o hija que va a desarrollar la EH.
Las mutaciones por expansión de segmentos de trinucleótidos se denominan dinámicas o inestables, ya que tienden a aumentar de una generación a la siguiente. El tamaño de la expansión de la secuencia CAG se correlaciona con la edad de inicio y gravedad de la enfermedad. De este modo, a mayor tamaño de la expansión, la enfermedad se inicia en edades más tempranas y es de progresión más rápida, y viceversa; explicándose así con el análisis molecular el fenómeno de la anticipación genética 16.
Dicha anticipación es mayor si la enfermedad la transmite un varón (imprinting o impronta genética). El tamaño de las expansiones es particularmente inestable en los espermatozoides y probablemente la meiosis repercute en gran medida en su inestabilidad, provocando aumento del número de repeticiones CAG, lo que explica que la transmisión paterna es la que provoca, con mayor frecuencia, el fenómeno de la anticipación. Dentro del propio Sistema Nervioso Central (SNC) la longitud de las expansiones es diferente según la región considerada; así, en el estriado y la corteza cerebral las expansiones CAG son más largas que en la corteza cerebelosa 17.
Anatomía patológica y cambios neuroquímicos
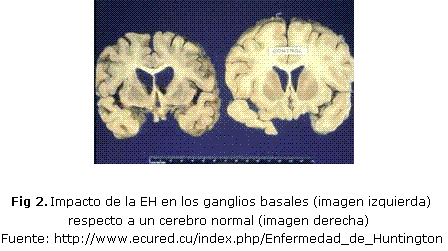
El cerebro presenta una atrofia cortical en relación directa con el grado de evolución de la enfermedad. En los cortes coronales el hallazgo característico es la atrofia del estriado, fundamentalmente del núcleo caudado, lo que condiciona un incremento en el tamaño de las astas frontales de los ventrículos laterales al perder la impronta de la cabeza del núcleo caudado (fig. 2). A diferencia de lo que ocurre en las enfermedades degenerativas ya desarrolladas, en la EH existe una lesión microscópica característica. El proceso degenerativo recae muy selectivamente en las neuronas estriatales espinosas de mediano tamaño 18-20.
Neuroquímicamente los niveles de sustancias neurotransmisoras como GABA y su enzima sintética descarboxilada del ácido glutámico están marcadamente disminuidos en los ganglios basales. Los niveles de acetilcolina, sustancia P y encefalinas también se encuentran reducidas. La espectroscopia por resonancia magnética en personas vivas afectadas muestra niveles elevados de lactato en los ganglios basales 21.
Las interneuronas estriatales, que carecen de espinas dendríticas, no están afectadas por el proceso degenerativo, y los neurotransmisores que manejan estas interneuronas (acetilcolina, somatostatina y neuropéptido) se encuentran preservados 22-23.
Fisiopatología
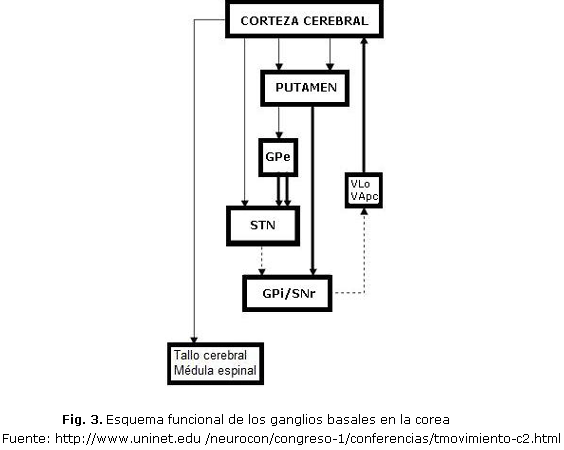
La EH se produce como consecuencia de una inactivación funcional o de una lesión en el NST (núcleo subtalámico), que conduce a una disminución de la actividad del complejo GPi/SNpr (globo pálido interno/sustancia nigra pars reticulata) (fig. 3).
La presencia de discinesia se vincula al hecho de que el proceso degenerativo en las neuronas del estriado se inicia en la subpoblación GABA-encefalina, conduciendo a una reducción del output inhibitorio en el circuito indirecto, lo que da lugar a una inhibición excesiva del NST por el GPe (globo pálido externo). Al disminuir el efecto excitador del NST sobre el complejo GPi/SNpr, disminuye el efecto inhibidor de éste sobre el tálamo, lo que conduce a un aumento de la actividad talamocortical y, en último término, a la aparición de movimientos involuntarios. Durante el proceso neurodegenerativo la progresiva afectación del circuito directo (subpoblación GABA-sustancia P) explica la disminución de la corea y la aparición paradójica de acinesia durante el curso de la enfermedad 7-8.
Manifestaciones clínicas
La EH se puede identificar clínicamente en presencia de: alteraciones del comportamiento, alteraciones afectivas y cognitivas asociadas a una disfunción motora progresiva y antecedentes familiares compatibles con una transmisión autosómica dominante 9.
La edad media de inicio de los síntomas es 38 años, con unos límites que pueden variar entre la segunda y la séptima décadas de la vida, pues como se expresó anteriormente la edad de inicio y gravedad de la enfermedad dependen del tamaño de la expansión de la secuencia CAG 9,21-22.
De forma característica los signos iniciales son la inquietud generalizada, alteraciones del sueño, cambios en el comportamiento, así como ansiedad y depresión 9.
Trastornos motores: al inicio los movimientos anormales son muy sutiles, integrados en el seno de los movimientos voluntarios o aparecen espontáneamente sin relación con estos. A medida que avanza la enfermedad los trastornos motores se hacen más prominentes, no son suprimibles y causan trastorno funcional. Con la evolución de la enfermedad la corea empeora afectándose el equilibrio de la marcha y los movimientos voluntarios. En estadios avanzados puede aparecer parkinsonismo, a menudo por el tratamiento anticoreico, así como posturas distónicas. El lenguaje se torna disártrico por las hipercinesias, siendo la comprensión normal. La hiperreflexia es común y en el 10% de los casos hay signo de Babinski.
En dependencia de la edad de inicio se han establecido tres formas clínicas. En la forma clásica la sintomatología comienza en la mayor parte de los pacientes entre los 20 y los 45 años de edad, generalmente entre 35 y 45, comportándose como explicamos anteriormente. La EHJdefinida por un comienzo antes de los 21 años de edad, representa alrededor del 10% de los pacientes con EH. Estos casos de comienzo precoz suelen caracterizarse por el predominio de un trastorno motor de carácter rígido-acinético-distónico (variante de Westphal), mayor deterioro intelectual y evolución más dramática de las manifestaciones clínicas.
Las crisis epilépticas son más frecuentes en la EHJ. Los síntomas psiquiátricos, la depresión y la psicosis son también frecuentes al inicio y durante la enfermedad. La forma senil, que inicia después de los 55 años se caracteriza por una corea pura, sin deterioro intelectual y progresión más lenta. La EH cursa además con otras alteraciones motoras como trastornos de la motilidad ocular voluntaria, que van desde el enlentecimiento de los movimientos oculares sacádicos hasta una dificultad severa de la motilidad ocular voluntaria. La impersistencia motora se manifiesta por la incapacidad para mantener los brazos extendidos durante algunos segundos o la lengua protruida durante 20 segundos o por un apretón de manos en el que el paciente no sostiene la contracción de la mano, sino que aprieta y suelta de forma repetitiva y ondulante (signo del apretón del lechero). Otros trastornos frecuentes en fases avanzadas de la enfermedad son la incoordinación motora, la ataxia, los trastornos del lenguaje y de la deglución, así como incontinencia urinaria en fase terminal 9,15,24-25.
Trastornos psiquiátricos: muchas veces constituyen el primer síntoma de la EH y hacen que los enfermos sean recluidos en centros psiquiátricos, incluyen cambios de personalidad, trastornos afectivos (depresión y menos frecuentemente manía), ilusiones y alucinaciones, paranoia y cuadros esquizofreniformes, agitación o apatía, disminución de la libido, inatención para seguir conversaciones, descuido del aseo personal y trastornos del sueño como somnolencia diurna e insomnio nocturno. Es frecuente la tendencia al suicidio 9,15,25.
Trastornos cognitivos: inicialmente consisten en alteración de la memoria reciente y el juicio, hasta desarrollar demencia que lleva a la incapacidad para realizar las actividades de la vida diaria. La demencia es de tipo “subcortical”, con predominio de bradifrenia (enlentecimiento del pensamiento), déficit de atención y de funciones ejecutivas con ausencia de alteraciones corticales como afasia, apraxias y agnosias 9,15,25
Diagnóstico nosológico y diferencial
El diagnóstico se basa en los datos clínicos y la comprobación de una transmisión vertical de herencia autosómica dominante. Las pruebas de neuroimagen (IRM y TC) ponen de manifiesto atrofia de la cabeza del núcleo caudado y de la corteza cerebral. La confirmación de la enfermedad, así como el diagnóstico prenatal y presintomático (estos dos últimos aún son temas de debate dado que estos aspectos no está exento de problemas legales y éticos) puede efectuarse mediante técnicas de genética molecular que demuestran la expansión patológica del triplete CAG en el gen IT15 26-27.
El diagnóstico diferencial de la EH se debe realizar con las otras causas de corea hereditaria y también de causas adquiridas, entre ellas:
Causas de corea hereditaria
- Enfermedad de HuntingtonHDL 1 y otras enfermedades por priones hereditariasHDL 2HDL 3Ataxias espinocerebelosas : SCA 17 (HDL 4), SCA 1-3Atrofia dentatorrubropalidoluysianaNeuroacantocitosis: corea-acantocitosis y síndrome de McLeod Corea hereditaria benignaNeurodegeneración con cúmulo cerebral de hierroEnfermedad de Wilson Ataxia de Friedreich
- Enfermedades mitocondriales
Causas adquiridas
-
Patología focal (enfermedad cerebrovascular y lesiones ocupantes de espacio)Coreas de base inmune: corea de Sydenham, lupus eritematoso sistémico y síndrome antifosfolipídico primario, corea gravídica, corea inducida por anticonceptivos orales y síndromes paraneoplásicos Corea de causa infecciosa: virus de inmunodefiencia humana, nueva variante de la enfermedad de Creutzfeldt-Jakob, tuberculosisEncefalopatías tóxicas y metabólicas: degeneración hepatolenticular crónica adquirida (no wilsoniana), hiperglucemia no cetósica, hipertiroidismo y policitemia rubra vera
-
Antiepilépticos: fenitoína, carbamazepina, valproato, gabapentina
El tratamiento debe ser multidisciplinario y aportar apoyo no solo al paciente sino también a sus familiares, por lo que es recomendable que intervengan asistentes sociales, genetistas, psicólogos y enfermeras conocedoras de la problemática de estos pacientes, además del neurólogo.
Aunque no existen tratamientos eficaces para ralentizar la progresión o retrazar el comienzo de la EH pueden y deben ser tratados los síntomas como la corea la depresión y la ansiedad 28-29.
Los únicos fármacos que se han demostrado eficaces en el control de la corea son los antagonistas de los receptores dopaminérgicos del cuerpo estriado (butirofenonas y neurolépticos) y los inhibidores del almacenamiento o liberación de la dopamina (tetrabenazina y reserpina) (tabla) 30-31.
Tabla . Fármacos usados para el tratamiento de la enfermedad Huntington
| Clase | Fármacos | Dosis inicial diaria | Dosis máxima diaria |
| Antipsicóticos típicos | Haloperidol | 0,5 mg | 8 mg |
| Flufenazina | 0,5 mg | 8 mg | |
| Tioridazina | 10 mg | 100 mg | |
| Tiotixene | 1 mg | 20 mg | |
| Antipsicóticos atípicos | Quetiapina | 12,5 mg | 100 mg |
| Clozapina | 2,5 mg | 100 mg | |
| Olanzapina | 2,5 mg | 30 mg | |
| Risperidona | 0,5 mg | 6 mg | |
| Ziprasidona | 20 mg | 160 mg | |
| Aripriprazol | 5 mg | 30 mg | |
| Fármacos depletores de dopamina | Tetrabenazina | 12,5 mg | 100 mg |
| Reserpina | 0,1 mg | 3 mg | |
| Benzodiacepinas | Clonazepam | 0,5 mg | 3 mg |
| Diazepam | 1 mg | 20 mg | |
| Alprazolam | 0,25 mg | 4 mg | |
| Antagonistas del glutamato | Amantadita | 100 mg | 500 mg |
Si bien los bloqueadores de la dopamina son moderadamente eficaces en la corea, pueden agravar la bradicinesia y la distonía. Los antipsicóticos atípicos como la clozapina, la risperidona y la olanzapina son mejor tolerados, pero no tan eficaces. Las indicaciones para el tratamiento de la corea incluyen interferencia en las actividades cotidianas y vergüenza social 32.
No existe tratamiento eficaz para la demencia, el aspecto más invalidante de la enfermedad. El donepezilo, una droga procolinérgica que es usada en el tratamiento de la enfermedad de Alzheimer, es inefectiva en el tratamiento de las alteraciones cognitivas producidas por la EH 32.
La depresión responde al tratamiento antidepresivo estándar. El tratamiento debe vigilarse cuidadosamente ya que puede producir manía o desencadenar suicidio, un problema muy grave en la EH. La ansiedad responde a la administración de benzodiazepinas y al tratamiento eficaz de la depresión. Se recomiendan las benzodiazepinas de acción prolongada más que las de acción breve, debido a su menor potencial de abuso y excitación paradójica. Se ha propuesto la utilidad de la coenzima Q10 (coQ10), isoniacida, muscinol y creatina, medicamentos aún en fase de evaluación 9,32-33.
En esta enfermedad es fundamental un consejo genético apropiado. La detección de los portadores de la enfermedad, en fases presintomáticas, es posible gracias a las nuevas técnicas de biología molecular; dado que la aplicación de éstas no está exenta de problemas legales y éticos (anteriormente explicado), en algunos lugares, se crea comités en los que participan enfermos, médicos, juristas y expertos en ética médica con el fin de asesorar en cada caso sobre la conveniencia de ponerlas en práctica.
En cuanto a la rehabilitación, el equipo de rehabilitación neurológica trabaja con el paciente y su familia, el cual contribuye a establecer objetivos de recuperación a corto y largo plazo.
Los objetivos de un programa de rehabilitación neurológica suelen ayudar al paciente a recuperar el máximo nivel posible de funcionalidad e independencia y a mejorar su calidad de vida general tanto en el aspecto físico, como en los aspectos psicológico y social. Un programa típico de rehabilitación neurológica ayuda a 34:
-
Realizar las actividades cotidianas como comer, vestirse, bañarse, ir al baño, escribir a mano, cocinar y las tareas básicas de la casa. Terapia del lenguaje: ayudarle a expresar sus ideas, su forma de hablar, la dicción y la comunicación. Asesoría (para combatir la angustia y la depresión). Actividades para mejorar el control y el equilibrio de los músculos del tronco, la pelvis y la cintura escapular. Un programa de ejercicios para mejorar la funcionalidad, seguridad y eficacia de los movimientos, para evitar o posponer la debilidad causada por la falta de uso, para controlar los espasmos y el dolor, para mantener la amplitud de los movimientos, y para desarrollar al máximo las capacidades potenciales de los músculos, los huesos y la respiración. Rehabilitación social Rehabilitación de la marcha y el equilibrio Asesoría nutricional Participación en los grupos de apoyo de la comunidad Actividades para mejorar los problemas cognoscitivos, como por ejemplo las dificultades de concentración, atención, memoria y juicio Educación con respecto a la enfermedad y su proceso
- Establecimiento de objetivos (a corto y largo plazo) contando con la participación del paciente y su familia
La rehabilitación vocacional no suele dar resultados satisfactorios por la dificultad de aprender tareas nuevas.
En cuanto al tratamiento quirúrgico en la EH se ha estudiado la cirugía del globo pálido (palidotomía y estimulación cerebral profunda) y los trasplantes fetales, sin obtener resultados 8.
Pronóstico
La enfermedad progresa lentamente y produce la muerte 15-20 años después del inicio de los síntomas. Las causas principales de muerte son: la neumonía por broncoaspiración secundaria a la disfagia y la inanición. Las formas juveniles evolucionan a mayor velocidad. No hay tratamientos efectivos para evitar la progresión de la enfermedad. La terapéutica se encamina al tratamiento sintomático de los problemas que surgen a lo largo de la evolución de dicha entidad 28,34.
REFERENCIAS BIBLIOGRÁFICAS
1. Fahn S, Jankovic J. Huntington disease. En: Principles and practice of movement disorders. Philadelphia: Editorial Churchil livingstone Elsevier; 2007.p.369-85.
2. Wild EJ, Tabrizi SJ. The differential diagnosis of Chorea. Practical Neurol. 2007; 7:360-373.
3. Arbor A, De Jong RN. George Huntington (1850-1916). En: The Founders of Neurology. 2ª ed. Illinois: Ed. Springfield Charles C Thomas Pub; 1970.p. 453-6.
4. Walker FO. Huntington’s disease. Lancet. 2007; 369: 218-28.
5. Vázquez Sánchez F, Rodríguez Martínez E, Arés Luque A. Actualización en coreas. Rev Neurol. 2009;48: 11-16.
6. Biglan KM, Shoulson I. Huntington’s disease. En: Hallett M, Poewe W. Therapeutics of Parkinson’s Disease and Other Movement Disorders. Chicester: Editorial John Wiley & Sons; 2008.p.295–315.
7. Velázquez Pérez L, Rodríguez Labrada R. Características generales de las enfermedades poliglutamínicas. En: Manifestaciones tempranas de la Ataxia espinocerebelosa tipo 2. Holguín: Ediciones Holguín; 2012.p.11-32.
8. Huntington's disease Collaborative Research Group. A novel gen containg a trinucleotide repeats that is expanded and instable on Huntington's disease chromosoma.Cel.1993; 72:971-83.
9. Azzarelli A. Enfermedad de Huntington y degeneraciones de algunos núcleos subcorticales. En: Neuropatología, diagnóstico y clínica. Barcelona: Editorial Edisma; 2000.p. 603-20.
10. Landles C, Bates G. Huntingtin and the molecular pathogenesis of Huntington disease. EMBO. 2004;5:958-963.
11. Kremer B, Almqvist E, Theilmann J. Sex-dependent mechanisms for expansions and contractions of the CAG repeat on affected Huntington disease chromosomes. Am J Hum Genet.1995; 57:343–350.
12. Wexler NS, Lorimer J, Porter J. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. Proc Natl Acad Sci. 2004;101: 3498–3503.
13. Brocklebank D, Gayan J, Andresen JM. Repeat instability in the 27–39 CAG range of the HD gene in the Venezuelan kindreds: counseling implications. Am J Med Genet B Neuropsychiatr Genet.2009;150:425–429.
14. Rosales Reynoso MA, Barros Núñez P. Biología molecular y Medicina. Diagnóstico molecular de la enfermedad de Huntington. Gac Méd Méx. 2008; 144(3):271-3.
15. Potter NT, Spector EB, Prior TW. Technical standars and guidelines for Huntington disease testing. Genet Med. 2004; 6:61-65.
16. Ravina B, Romer M, Constantinescu R. The relationship between CAG repeat length and clinical progression in Huntington’s disease. Mov Disord. 2008; 23:1223–1227.
17. Ross RA, Vegtervan der Vlis M, Hermans J, Elshove HM, Moll AC, Van de Kamp JJ, et al. Age at onset in Huntington's disease: effect on line of inherintance and patient's sex. Nat Genet.1993; 28:515-9.
18. Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP. Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol. 1985; 44:559–577.
19. Vonsttel JP, Keller C, Del Pilar Amaya M. Neuropathology of Huntington’s disease. En: Vinken PJ, Bruyn GW. Handbook of Clinical Neurology. Amsterdam: Editorial Elsevier; 2008.p. 599–618.
20. Aucoin JS, Jiang P, Aznavour N. Selective cholinergic denervation, independent from oxidative stress, in a mouse model of Alzheimer’s disease. Neuroscience. 2005; 132:73–86.
21. Penney JB, Young AB. Striatal inhomogeneities and basal gan-glia function. Mov Disord. 1986;1:3–15.
22. Albin RL, Qin Y, Young AB, Penney JB, Chesselet MF. Preproenkephalin messenger RNA-containing neurons in striatum of patientswith symptomatic and presymptomatic Huntington’s disease: an in situ hybridization study. Ann Neurol.1991; 30:542–549.
23. Albin RL, Reiner A, Anderson KD. Preferential loss of striato-external pallidal projection neurons in presymptomatic Huntington’s disease. Ann Neurol. 1992;31:425–430.
24. Jankovic J. Movement Disorders. En: Bradley WG, Daroff RB, Fenichel GM, Jankovic J, Mazziotta JC. Bradley’s Neurology in Clinical Practice. Philadelphia: Editorial Elsevier Saunders; 2012.p. 1762-1801.
25. Ropper AH, Samuels A. Degenerative Diseases of the Nervous System En: Adams & Victors' Principles of Neurology. 9 ed. Philadelphia: Ed. McGraw-Hill; 2009.p.895-954.
26. Tibben A, Duivenvoorden HJ, Vegter-van der Vlis M. Presymptomatic DNA testing for Huntington disease: identifying the need for psychological intervention. Am J Med Genet. 1993; 48:137–144.
27. Wild EJ, Mudanohwo EE, Sweeney MG, Schneider SA, Beck J, Bhatia KP, et al. Huntington’s disease phenocopies are clinically and genetically heterogeneous. Mov Disord. 2008; 23: 716-20.
28. Hersch SM, Rosas HD. Neuroprotection for Huntington’s disease: ready, set, slow. Neurotherapeutics. 2008;5: 226-36.
29. Mestre T, Ferreira J, Coelho MM, Rosa M, Sampaio C. Therapeutic interventions for disease progression in Huntington’s disease. Cochrane Database Syst. 2009[citado 25 jun 2013]; 8(3) Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/19588392
30. Huntington Study Group. Tetrabenazine as antichorea therapy in Huntington disease: a randomized controlled trial. Neurol. 2006; 66:366–372.
31. Adam OR, Jankovic J. Symptomatic treatment of Huntington disease. Neurotherapeutics. 2008; 5: 181-97.
32. Hyson HC, Kieburtz K, Shoulson I. Safety and tolerability of high-dosage coenzyme Q10 in Huntington’s disease and healthy subjects. Mov Disord. 2010;25:1924–1928.
33. Shoulson I, Young AB. Milestones in Huntington Disease. Mov Disord. 2011; 26(6):1127-1133.
34. Discapnet. (sede web). Enfermedad de Huntington. España: Fundación ONCE; 2009 [citado 27 jun 2013]. Disponible en:http://salud.discapnet.es/Castellano/Salud/Discapacidades/Discapacidades%20Neurologicas/Enfermedad%20de%20Huntington/Paginas/Cover%20corea.aspx
Recibido: 2 de julio de 2013
Aprobado: 19 de julio de 2013
Dr. Jorge Michel Rodríguez Pupo. Hospital General Universitario Vladimir Ilich Lenin. Holguín. Cuba.
Correo electrónico: yuna@hvil.hlg.sld.cu
