










 Serviços customizados
Serviços customizados Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
Los compuestos orgánicos persistentes (COP) son sustancias tóxicas creadas por el hombre que presentan un tiempo de residencia elevado en el medio ambiente debido, fundamentalmente, a su elevada resistencia a la degradación química y biológica, y al hecho de que han sido vertidos al entorno en cantidades que superan la capacidad del medio natural para degradarlos. 1 Específicamente los plaguicidas organoclorados tales como la clordecona (CLD, C10Cl10O, CAS: 143-50-5) y el β-hexaclorociclohexano (β-HCH, C6H6Cl6, CAS 319-85-7) son de gran preocupación pues fueron ampliamente utilizados alrededor del mundo, 2-4 presentan una elevada estabilidad en el medio ambiente y causan severas afectaciones a la salud humana, los animales y el medio ambiente en general. Debido a esto, los mismos fueron incluidos desde 2009 en la lista de COP por la Convención de Estocolmo. 5
En los últimos años se ha promovido la búsqueda de estrategias para la descontaminación de aguas centradas en plaguicidas organoclorados. 6-8 Los métodos de descontaminación utilizados van desde procesos avanzados de oxidación, 9 a el uso de carbones activados para el tratamiento de aguas contaminadas mediante adsorción. 10-13 A pesar de estos esfuerzos todavía resulta necesario incrementar la eficiencia de los métodos empleados, lo que ha propiciado la búsqueda de nuevas alternativas como la formación de complejos de inclusión molecular con ciclodextrinas. 14
Las ciclodextrinas (CD) son una familia de oligosacáridos cíclicos consistentes en un número de subunidades α-D-glucopiranosa enlazadas entre sí mediante enlaces glicosídicos (1→4). Las CD más comunes, llamadas α-, β- y γ-CD, constan respectivamente de 6, 7 y 8 unidades glucopiranosas. 15 Las destacadas capacidades de encapsulación de las CD causan un tipo de interacción anfitrión-huésped 16-18 que ha permitido su empleo en aplicaciones para muchos sectores de interés para la sociedad como la agricultura o el medio ambiente. 19,20 Particularmente interesante para los propósitos de esta investigación debe resaltarse su uso en alternativas innovadoras para la remoción de contaminantes de aguas y suelos, basado en la formación de complejos de inclusión insolubles en agua y la mayoría de los solventes orgánicos. 19,21-23 Recientemente, Rana y col. 14 demostraron que la ciclodextrina es un buen agente acomplejante para la remoción de clordecona de las aguas contaminadas, formando complejos de inclusión 1:1 con la β- y γ-CD.
La determinación de CLD y β-HCH en agua representa un problema analítico debido a las bajas concentraciones de estos plaguicidas en la naturaleza, las cuales en ocasiones se encuentran por debajo de los límites de cuantificación de muchos métodos analíticos modernos. El empleo de métodos radioanalíticos constituye una alternativa para disminuir los límites de cuantificación, puesto que estos métodos son muy sensibles. El marcaje de la CLD y el β-HCH con un isótopo radiactivo de yodo puede producir radiotrazadores análogos a estos plaguicidas. Estos radiotrazadores podrían usarse entonces para estudiar el proceso de descontaminación, en concentraciones similares a las observadas para la CLD y el β-HCH en una planta de tratamiento de agua.
La modelación molecular de estos sistemas puede ser una alternativa económica y limpia para estudiar el proceso de inclusión molecular, como ha sido comprobado en el caso de varios COP para los estudios de adsorción en carbón activado. 24-27 Además, la modelación teórica es una herramienta que permite la descripción detallada del proceso de asociación a nivel molecular, así como también debe ser el primer paso a realizar para la evaluación del desempeño de radiotrazadores análogos a la CLD y el β-HCH en el proceso de inclusión molecular.
El objetivo general de esta investigación es evaluar la formación y estabilidad de los posibles nanoagregados plaguicidas@CD caracterizando las interacciones de la CLD, el β-HCH y sus análogos marcados con las CD naturales.
MÉTODOS
Sistema objeto de estudio
Para una evaluación más rigurosa de las interacciones entre las CD y los plaguicidas de interés (CLD y β-HCH), se estudiaron diferentes confórmeros de cada CD. Se utilizó un grupo de 24 confórmeros simétricos, 8 de cada CD. Estos confórmeros fueron caracterizados por Gamboa-Carballo y col. 28 y difieren en la orientación de los patrones de enlaces de hidrógeno intramoleculares. La figura 1 muestra una representación de los 8 confórmeros para la α-CD.

Fig. 1 Confórmeros simétricos de las CD. Para mayor claridad, en cada confórmero solo se presentan la α-CD y se desataca una única unidad glucopiranosa.
Para el estudio de los complejos con ICLD y I-β-HCH se utilizaron solo los confórmeros tipo B y C de las 3 ciclodextrinas estudiadas. La elección de estos confórmeros se basó en resultados obtenidos para la CLD y el β-HCH que serán discutidos en los resultados de esta investigación.
Detalles computacionales
Para la realización del estudio se procedió a realizar primeramente un estudio del espacio de las interacciones CLD@CD, β-HCH@CD, ICLD@CD e I-β-HCH@CD por la metodología de las Hipersuperficies de múltiples mínimos (MMH). 29 Dicha metodología combina métodos cuánticos semiempíricos para la evaluación de las energías con mecánica estadística para obtener propiedades termodinámicas relacionadas con la asociación molecular. 13,25,30) La energía de asociación se define como
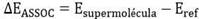
dónde,
 es la energía del complejo molecular formado por la CD y el plaguicida o su análogo marcado mientras que
es la energía del complejo molecular formado por la CD y el plaguicida o su análogo marcado mientras que
 es la suma de las energías de cada una de las moléculas independientes. Una asociación termodinámica favorable implica que la supermolécula será más estable que las moléculas aisladas. 13 A pesar de que el
es la suma de las energías de cada una de las moléculas independientes. Una asociación termodinámica favorable implica que la supermolécula será más estable que las moléculas aisladas. 13 A pesar de que el
 es el criterio termodinámico utilizado para definir la espontaneidad de un proceso en este trabajo se utilizó el
es el criterio termodinámico utilizado para definir la espontaneidad de un proceso en este trabajo se utilizó el
 debido a que este criterio termodinámico presenta un comportamiento similar por lo que ha sido usado en estudios anteriores. (11,13
debido a que este criterio termodinámico presenta un comportamiento similar por lo que ha sido usado en estudios anteriores. (11,13
El procedimiento MMH estándar coloca al soluto en el centro de una caja cúbica y entonces genera dentro las moléculas del diluyente en configuraciones aleatorias 31 utilizando el programa Granada. En nuestro caso se ubicó la molécula anfitriona (las CD) en el centro de la caja y el contaminante se colocó en configuraciones aleatorias en el interior de la caja.
El Hamiltoniano semiempírico PM6-D3H4X 32-35 se utilizó para la optimización de 200 geometrías y energías diferentes para cada sistema, implementadas en el software MOPAC2016. 36 PM6 es un Hamiltoniano semiempírico moderno que mejora la descripción de los enlaces por puente de hidrógeno con respecto a los métodos semiempíricos precedentes. 32 Para una descripción más precisa del sistema de estudio se le introdujeron 2 correcciones, la primera D3H4 34,35 integra correcciones avanzadas a las interacciones dispersivas y por puente de hidrógeno. Mientras la segunda X 33,35 rectifica el fallo presente en PM6 a la hora de describir las interacciones de los halógenos (Cl, Br, I) con átomos más electronegativos que ellos como O y N pues el conjunto de funciones base usado no permite describir la anisotropía de la densidad electrónica en los átomos de halógenos (hueco-σ), responsables de la contribución a la atracción electrostática en el enlace de los halógenos con los átomos antes mencionados (O y N).
A continuación, las estructuras representativas de los diferentes tipos de interacciones fueron reoptimizadas usando la teoría del funcional de la densidad (DFT). 37,38 Para la reoptimización de las estructuras distintivas a nivel de DFT se utilizó, con la base de Pople 6-31G(p,d), un funcional híbrido que utiliza la aproximación del gradiente meta-generalizado: M06-2X 39,40 y que permite describir correctamente las energías de enlace de dímeros no enlazados covalentemente 40 siendo adecuados para la descripción de interacciones de Van der Waals como las presentes en nuestro sistema. Los efectos del solvente fueron tenidos en cuenta a partir del modelo de solvente implícito SMD. 41) Las energías obtenidas fueron corregidas para mitigar el error de superposición de bases (BSSE por sus siglas en inglés) usualmente presente en estos cálculos mediante un método desarrollado por los autores.
Posteriormente para una mejor descripción de la densidad electrónica, la función de onda se optimizó utilizando la base de Pople 6-311+G(2df,2pd). Todos los cálculos se realizaron con el programa Gaussian 09. 42 Las estructuras representativas de los complejos CLD@CD y β-HCH@CD se estudiaron utilizando la teoría de átomos en moléculas (QTAIM), 43,44 propuesta por Bader y col., con el objetivo de caracterizar los tipos de interacciones predominantes en dichos complejos de inclusión a partir de los criterios de Nakanishi 45,46 permitiendo analizar la naturaleza de estas interacciones. Un procedimiento análogo se utilizó para los complejos con ICLD e I-β-HCH con la diferencia de que el pseudopotencial LANL2DZ 47 se utilizó para el átomo de yodo. Todos los cálculos QTAIM se realizaron a partir de los ficheros de funciones de onda generados por Gaussian09 42 con el programa Multiwfn 3.3.6. 48
Mitigación del error de superposición de base
En sistemas como el que nos ocupa, el BSSE tiende a ser importante. Por un lado, la cantidad elevada de electrones a describir en el sistema de estudio nos obliga a limitar el tamaño de la base a utilizar. Un aumento del tamaño de la base implicaría una disminución del BSSE y, al mismo tiempo, un aumento considerable del tiempo de cálculo lo que hace casi impracticable esta medida más allá de una base 6-31G(d,p) para el estudio de estos complejos. Además, la gran cantidad de interacciones no covalentes esperadas en las supermoléculas a estudiar incrementa el BSSE proporcional al número de electrones involucrados en estas interacciones. 49
En el presente trabajo se presenta, por primera vez, una modificación al método de contrapeso para la mitigación del BSSE que se basa en la idea de aprovechar la propiedad de la energía de asociación de ser una función de estado para calcular la energía de interacción en fase gaseosa, mientras se utiliza un ciclo termodinámico para desolvatar y resolvatar el sistema calculado. La figura 2 muestra el esquema de la idea original basada en un ciclo termodinámico que conlleva al mismo resultado de energía independientemente del camino escogido para calcularla.
Como se explicó anteriormente, los procesos concertados de deformación e interacción presentes en la asociación de los componentes del sistema AB, se pueden tratar de manera independiente, ya que la energía es una función de estado. Del mismo modo, la desolvatación y resolvatación arbitraria y ficticia del sistema no afecta el cálculo de la energía de interacción, cuyo BSSE es impracticable corregir en E3, pero fácilmente corregible en E6.
La ecuación 1 muestra los cálculos de energía a realizar para lograr la corrección de E0. Aquí E1 y E2 son las energías de deformación de los componentes A y B respectivamente y E3 es la energía de formación del complejo AB en fase acuosa y, en consecuencia, de interacción de los reaccionantes.

Fig. 2 Esquema general del ciclo termodinámico generado para el cálculo de la energía de asociación (E0). E1 y E2 son las energías de deformación de los componentes del sistema separados en el infinito. E4, E5 y E7 son las energías de solvatación de los componentes y la supermolécula, respectivamente. E3 es la energía de interacción de los componentes en fase acuosa (ac) y E6 es la misma en fase gaseosa (g). El subíndice “def” indica deformación.
E3 no se puede corregir usando el método de contrapeso y se calcula mediante la ecuación 2 siguiendo el esquema de la figura 2.

donde E4, E5 y E7 son las energías de solvatación de A, B y AB, respectivamente. La energía de interacción en fase gaseosa E6 responsable de la formación del complejo “AB” puede ser fácilmente corregida usando el método de contrapeso, por lo que la energía de asociación quedaría más correctamente escrita según la ecuación 3.
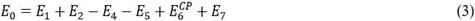
donde el superíndice “CP” de E6 indica que esta energía de interacción está corregida usando el método de contrapeso.
El sistema de ecuaciones 4 muestra la simplificación del problema de cálculo, reduciendo la complejidad del esquema presentado en el esquema de la figura 2 y la ecuación 3.
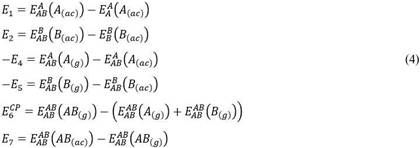
De esta manera obtenemos una nueva expresión para la energía de asociación (E0) de acuerdo la ecuación 5 y al esquema de la figura 3.

Este esquema de cálculo nos permite corregir el BSSE de un sistema supramolecular teniendo en cuenta los efectos del solvente sobre la energía con un mínimo de gasto computacional. Todas las geometrías usadas en los cálculos en fase gaseosa corresponden exactamente con aquellas obtenidas teóricamente en disolución, por lo que no se necesita ninguna optimización de las geometrías y permite la corrección realizando cálculos puntuales de energía.

Fig. 3 Esquema final del ciclo termodinámico simplificado para el cálculo de la energía de asociación (E0). E1 y E2 son las energías de deformación de los componentes del sistema separados en el infinito. E6 es la energía de interacción de los componentes. Estas energías se calculan en conjunción con E4, E5 y E7 que son las energías de solvatación de los componentes y la supermolécula, respectivamente. El subíndice “def” indica deformación.
Estudio experimental de la formación de los nanoagregados
Preparación de los complejos β-HCH@ciclodextrinas
Las CD de grado analítico (> 98 %) se obtuvieron de Merck y Fluka. 50 El β-HCH se obtuvo de la Sigma Aldrich Chemical Company (St Louis). Se prepararon soluciones de 0,01 mM de α, β y γ-CD, respectivamente, en agua destilada, así como una disolución de β-HCH en cantidad estequiométrica mediante la disolución previa en una cantidad mínima de metanol. Luego de la solubilización en sus respectivos solventes, se mezclaron 10 mL de cada una las soluciones de las moléculas anfitrionas con 10 mL de la solución de β-HCH, y en aquellas en las que apareció un precipitado, indicando la formación de los complejos de inclusión (-HCH@CD, el sobrenadante se decantó y el sólido se lavó con una mezcla de agua-etanol (1:1). El precipitado obtenido se secó al vacío a temperatura ambiente en una desecadora sobre silicagel.
Cuantificación del (-HCH
La cuantificación del (-HCH en solución acuosa se llevó a cabo mediante cromatografía líquida (CL) acoplada a un espectrómetro de masas (AGILENT LC/MS serie del sistema 1100). La separación por CL se realizó usando una columna C8 (150 mm, Eclipse X08-C8) a 80 °C usando el siguiente gradiente de fuerza eleutrópica: 0-6 min una mezcla de 55 % de ACN en agua y un minuto adicional con 100 % ACN. La ionización del (-HCH se realizó mediante electronebulización en modo de ion negativo. Los parámetros finales de la cámara del nebulizador con un flujo de gas secante de 12 L∙min-1, a 350 °C, fueron una presión del atomizador: 35 psi, una tensión capilar: 4000 V, energía de colisión: 50 eV. El pico en el espectro de masa del ion molecular del (-HCH está en 507/509 (M/M+2).
Técnicas espectroscópicas de caracterización
Los espectros raman de barrido de las CD (α, β y γ-CD), β-HCH y las diferentes muestras de precipitados formados se midieron empleando un microscopio Horiba scientific LabRAM HR evolution con un haz láser de iones Ar+ de 514,5 nm. Los tiempos de acumulación fueron de 20 seg. Los espectros infrarrojos con transformada de Fourrier (FTIR) se colectaron usando espectrómetro Perkin Elmer spectrum one FTIR infrared spectrometer con un accesorio de muestreo de reflexión total atenuada (ATR, cristal compuesto de diamante/ZnSe), con una resolución espectral de 4 cm-1 para 8 mediciones.
RESULTADOS Y DISCUSIÓN
Nanoagregados de CLD y β-HCH con CD
Con el objetivo de evaluar el comportamiento químico de las CD un estudio teórico de su equilibrio conformacional se realizó por Gamboa-Carballo y col. 28 Se encontraron y caracterizaron, para cada una de las CD estudiadas, 8 confórmeros simétricos los cuales difieren en los patrones de enlaces de H intramoleculares. Los resultados obtenidos se encuentran en concordancia con los datos disponibles de difracción de rayos X lo cual contribuyó a validar los resultados de este trabajo. 51-53 Estos cálculos mostraron que 4 de los 8 confórmeros evaluados para cada CD son los que deben predominar en disolución acuosa.
Se empleó la metodología MMH para realizar una exploración exhaustiva del espacio de configuraciones y evaluar las propiedades termodinámicas de asociación que describen el proceso de inclusión molecular. Las figuras 4A y 4B muestran la energía de asociación media para los complejos CLD@CD 54 y β-HCH@CD 55 respectivamente.
Como puede apreciarse, existe una estabilización considerable de los nanoagregados cuando la γ-CD es la molécula anfitriona para todos los confórmeros evaluados. En ambos casos, ocurre una estabilización progresiva de los nanoagregados mientras aumenta el tamaño de la cavidad de las CD para los confórmeros B2, B3, C1 y C4. De estos confórmeros se seleccionaron, las estructuras representativas de los complejos de inclusión para un posterior refinamiento. Se seleccionaron, en todos los casos, el mínimo global y estructuras adicionales que resultaron de interés por las geometrías presentadas. Esta selección se llevó a cabo teniendo en cuenta que estas estructuras tuvieran una población mayor al 10 % de acuerdo a una distribución de Boltzmann.

Fig. 4 Energías de asociación medias (calculadas empleando la metodología MMH) de (a) CLD 54 y (b) β-HCH 55 con los confórmeros estudiados de las CD.
El refinamiento de la geometría de los complejos, sus energías de asociación y sus funciones de onda se realizó a través de cálculos DFT empleando el funcional híbrido meta-GGA M06-2X con la base de Pople 6-31G(d,p). Las energías de asociación aquí calculadas fueron corregidas para mitigar el BSSE empleando un método desarrollado por los autores. 54 Estos cálculos confirmaron que los complejos más estables se forman cuando la γ-CD es la molécula anfitriona. Los confórmeros B3 y C1 forman los nanoagregados termodinámicamente más estables para ambos plaguicidas.
Para la caracterización de las interacciones presentes en estos complejos se realizó un análisis topológico de la densidad electrónica basado en la QTAIM y usando los criterios de Nakanishi 45,46 para determinar el tipo de interacciones a partir de funciones dependientes de la densidad electrónica. A partir de este análisis fue posible concluir que el gran número de interacciones dispersivas (con un mínimo de 17 interacciones entre los contaminantes y la CD) unido a la presencia de otras interacciones de mayor fortaleza como enlaces de hidrógeno, enlaces dihidrógeno y enlaces de halógeno contribuyen a explicar la estabilidad de estos complejos a pesar de la ausencia de interacciones covalentes.
Los resultados teóricos obtenidos para los sistemas CLD@CD se encuentran en concordancia con los resultados experimentales obtenidos por Rana y col. en 2016, 14 mientras que aquellos de los complejos β-HCH@CD fueron confirmados mediante espectroscopía Raman y FT-IR y análisis de microscopía electrónica realizados por Ferino-Pérez y col. 55
Radiotrazadores del sistema a partir de análogos marcados
El trabajo de Jáuregui-Haza y col. 56 evalúo teóricamente la posibilidad de emplear análogos marcados con un isótopo radiactivo de yodo como trazadores del sistema objeto de estudio. Los resultados obtenidos en esta investigación nos permitieron verificar esta hipótesis. La exploración del espacio de configuraciones entre los plaguicidas marcados (ICLD y I-β-HCH) con los 4 confórmeros más estables en disolución acuosa de las 3 ciclodextrinas naturales mostró al ser comparado con los resultados mostrados en la figura 4 que ambos compuestos presentan una tendencia de asociación similar a la que exhibieron sus análogos no marcados. La figura 5 muestra la similitud existente en las geometrías e interacciones presentes cuando se comparan los complejos formados con los plaguicidas con aquellos formados con sus análogos yodados.
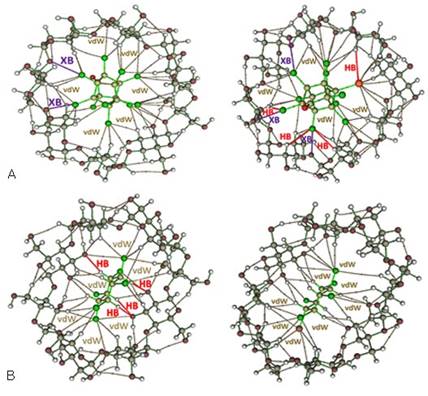
Fig. 5 Interacciones intermoleculares de los complejos de inclusión molecular (A) CLD@C1-(-CD e I-CLD@C1-(-CD y (B) (-HCH@C1-(-CD e I-(-HCH@C1-(-CD determinados mediante el análisis QTAIM de los valores de la función de onda obtenidos usando el esquema de cálculo M06-2X/6-31G(d,p). Los diferentes tipos de interacciones se identifican como: dispersivas (vdW, en amarillo), enlaces de hidrógeno (HB, en rojo) y enlace de halógeno (XB, en violeta).
Conclusiones
El presente estudio demuestra la formación de complejos de inclusión CLD@CD y β-HCH@CD estables, lo que corrobora la hipótesis de que es posible utilizar las CD en una amplia gama de alternativas para el tratamiento de aguas contaminadas con los plaguicidas estudiados. Los complejos más estables se forman cuando la γ-CD es la molécula anfitriona, con una mayor estabilización de las interacciones en aquellos que un mayor grado de oclusión (Confórmeros tipo C). El método desarrollado permite mitigar el BSSE cuando se emplean modelos de solvente implícito sin un aumento considerable del costo computacional. La introducción de sustituyentes electroaceptores en las CD puede aumentar la estabilidad de los complejos de inclusión formados. Los estudios QTAIM reflejan que las interacciones en los complejos de inclusión CLD@CD y β-HCH@CD son predominantemente de carácter dispersivo. La formación de los nanoagregados β-HCH@CD se comprobó experimentalmente. La posibilidad de utilizar la ICLD y el I-β-HCH como radiotrazadores de la CLD y el β-HCH en la formación de complejos de inclusión molecular con CD se demostró teóricamente.

