










 Serviços customizados
Serviços customizados Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroducción
Los enfoques de gestión del riesgo son de gran utilidad en las producciones médico-farmacéuticas y biotecnológicas que aplican las normas relacionadas con los dispositivos médicos, la Administración de Riesgos a la Calidad y las Buenas Prácticas de Producción Farmacéutica.1
Los dispositivos médicos (DM) están constituidos por los sencillos descartables, los implantables, los equipos electromédicos e incluso los software médicos y reactivos de diagnóstico in vitro.2 Una de sus funciones es la de diagnosticar enfermedades o lesiones,3 por tanto, la seguridad y la eficacia son requisitos esenciales que deben cumplir. Debido a su amplia gama, para cada uno de ellos se establecen precisiones, especialmente para los que tienen funciones de medición y los empleados para el diagnóstico clínico a través de ensayos.
Asimismo se establecen regulaciones estatales encaminadas a eliminar, o reducir a niveles aceptables, la ocurrencia de eventos adversos relacionados con el uso de los dispositivos médicos, o sea, se protege a la sociedad de su uso indebido y de productos dañinos.4,5 Estas disposiciones requieren que los fabricantes implementen y mantengan un Sistema de Gestión de la Calidad (SGC), según la norma NC-ISO 13485:2018,3 y que realicen un proceso de gestión de riesgos mediante la aplicación de la norma ISO 14971:2019 (fig. 1).1
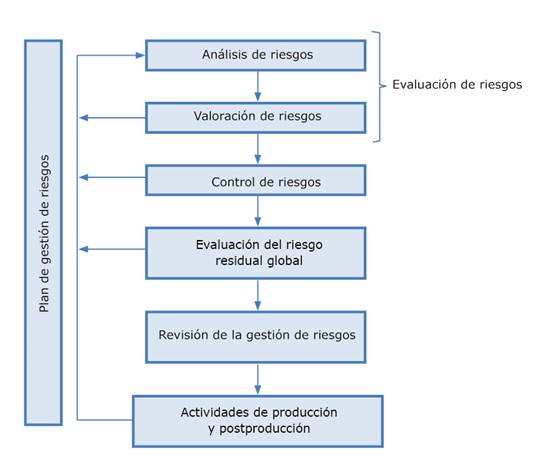
Los diseñadores y fabricantes de DM tienen el conocimiento científico-técnico para garantizar productos seguros y eficaces; pero no siempre disponen de la experiencia para gestionar los riesgos. Por consiguiente, el objetivo de este trabajo fue proponer una guía para la gestión de los riesgos indirectos en pacientes con diagnósticos incorrectos o retrasados.
Métodos
Para la elaboración de la guía se emplearon los documentos internacionales:
También se analizaron expedientes de gestión de riesgos de DM que evidenciaron el desconocimiento y la incorrecta interpretación de los documentos normativos de referencia.
Resultados
La Guía para la Gestión de Riesgos aplicada a dispositivos médicos para el diagnóstico (DMD) es un documento de apoyo para los investigadores del Centro de Gestión y Desarrollo de la Calidad de la Oficina Nacional de Normalización y el Centro de Biomateriales de la Universidad de La Habana.8 Aborda los riesgos indirectos generados en pacientes con diagnósticos incorrectos o retrasados. El concepto de riesgo posee dos componentes:7 la probabilidad de que ocurra un daño y la consecuencia de ese daño, o sea, su gravedad.
Cuando un fabricante suministra un DM, cualquier riesgo que no pueda ser controlado a través del diseño o las medidas de protección se transfiere a la institución que adquirió el equipo; por tanto, el DM debe tener la información de seguridad que controle esos riesgos residuales para que la institución médica pueda evaluarlos y determine si son aceptables. Aunque los fabricantes proporcionen información de seguridad sobre el DM, no pueden influir en las acciones de los especialistas que ordenan, reciben y actúan sobre los resultados del examen.
El DMD con función de medición debe diseñarse con medidas de control de riesgos adecuadas para los usuarios previstos y el entorno de uso especificado fuera de los laboratorios; por ejemplo, cuando se utiliza por los médicos o por los pacientes.
El análisis de riesgos es el uso sistemático de la información disponible para identificar los peligros y comprende cuatro etapas: uso previsto y mal uso razonablemente previsible, características relacionadas con la seguridad del paciente, identificación de peligros y situaciones peligrosas y estimación de riesgos. El peligro es una fuente potencial de daño, se conceptualiza como lesión o perjuicio a la salud de las personas, daños a la propiedad o al medio ambiente.1
Todo análisis de riesgos comienza por la descripción del DM y su función específica en el resultado del examen. Los DMD tienen un uso analítico y un uso clínico determinado por sus dos usuarios: el que realiza el examen y el que lo recibe, interpreta y actúa sobre los resultados.6
Se considera uso previsto aquel para el que está destinado un DM de acuerdo con las especificaciones, las instrucciones y la información proporcionadas por el fabricante. Sus elementos son la indicación médica prevista, la población de pacientes, la parte del cuerpo o el tipo de tejido con el que interactúa, el perfil del usuario, el entorno de uso y el principio de funcionamiento. En cambio, un mal uso razonablemente previsible es el empleo incorrecto de un equipo o sistema médico. Usarlo de la forma no prevista por el fabricante puede ser intencional o no, pero resulta del comportamiento de los tipos de usuarios.
Para caracterizar el DMD y sus usos, en la guía aparecen un conjunto de preguntas que pueden realizarse a partir del Anexo A del reporte técnico ISO/TR 24971:2020 Medical devices - Guidance on the application of ISO 14971.6
En cuanto a las características relacionadas con la seguridad del paciente, estas pueden ser de desempeño, confiabilidad o comunicación. En las de desempeño se analiza si los exámenes son cuantitativos, semicuantitativos o cualitativos.6 Cuando los exámenes son cuantitativos los resultados falsos altos, normales o bajos pueden afectar un diagnóstico, causar una terapia inapropiada o retrasada y provocar daños al paciente. El tipo y la gravedad del daño pueden depender de la magnitud del error en los puntos de decisión médica. Para los DMD cuantitativos, las características relevantes de su desempeño incluyen la exactitud (veracidad y precisión) de los resultados, la especificidad, la sensibilidad, los límites de detección y la cuantificación e intervalo de medición.
Los exámenes semicuantitativos proporcionan una aproximación clínicamente útil de la cantidad que se mide. Generalmente los valores se asignan en función de una escala ordinal o se informan como una cantidad límite (punto de corte). Para estas pruebas, las características de desempeño del DMD pueden incluir precisión, sensibilidad, especificidad y límite de detección.
Los exámenes cualitativos determinan la presencia o ausencia de un efecto y los resultados se informan como positivos, negativos o indeterminados. Los valores de corte y las bases de datos relevantes pueden definir resultados positivos o negativos. Un resultado positivo, cuando el analito está ausente (falso positivo), o un resultado negativo, cuando el analito está presente (falso negativo), pueden afectar el diagnóstico o el tratamiento. Las características de desempeño del DMD para este tipo de exámenes pueden incluir precisión, sensibilidad diagnóstica (fracción de resultados positivos verdaderos en pacientes con enfermedad), especificidad (fracción de resultados negativos verdaderos).
La confiabilidad del DMD se relaciona con la seguridad del paciente y depende de que los resultados de los exámenes se ofrezcan en el momento oportuno, cuando los médicos deben tomar decisiones urgentes. Las características de confiabilidad de los DMD pueden incluir fiabilidad del sistema y del software, compatibilidad de los componentes, uso del sistema y estabilidad de los reactivos y controles.
En caso de que se empleen tecnologías de la información digital hay otras características relacionadas con la seguridad del paciente; entre ellas, la correcta identificación del paciente, de la muestra y otras informaciones necesarias, las características del dispositivo que puedan conducir a la alteración o corrupción de datos en los procesos de recopilación, almacenamiento y reporte.
Las características de la tecnología de la información digital que pueden afectar la seguridad del paciente podrían ser las conexiones entre dispositivos o redes, la transmisión de datos por Internet, el interfaz con aplicaciones digitales, las aplicaciones de software integradas, la transferencia de datos sin blindaje, el almacenamiento de los datos digitales y la interrupción de otros dispositivos conectados.
El resultado de un examen diagnóstico se considera un peligro para el paciente si conlleva a6) una intervención médica inapropiada o a la no intervención médica necesaria para evitar daños. Las posibles eventualidades del uso de un DMD son resultado de un examen incorrecto o de un examen retrasado y la información errónea que acompaña a ese resultado.
Los riesgos específicos deben identificarse por la magnitud y dirección del error, la extensión del retraso o la información auxiliar incorrecta o ausente. Los peligros pueden ocurrir en condiciones de falla del DMD o en su uso normal. En la guía se ejemplifican las condiciones de falla y los peligros derivados del uso normal; ambos corresponden a las limitaciones inherentes a la tecnología del DMD según el Anexo H del reporte técnico ISO/TR 24971:2020 Medical devices - Guidance on the application of ISO 14971.6
Cada resultado de examen está sujeto a fuentes de variabilidad. Pueden ocurrir resultados inexactos por las siguientes causas:
las tasas inherentes de falsos negativos y falsos positivos de los procedimientos cualitativos causadas por la incertidumbre de los valores de corte asignados;
la incertidumbre de la medición asociada con los exámenes cuantitativos;
errores en la determinación estadística de los rangos normales;
influencia de factores o sustancias interferentes;
variabilidad biológica y características químicas del analito y la matriz;
la fiabilidad finita de los componentes del instrumento.
Otra fuente delicada son los errores de uso, tales como no considerar requisitos especiales fuera de la rutina del laboratorio; alterar la secuencia de las operaciones y errores de entrada de datos. Cuando los pacientes emplean el DMD para la autocomprobación pueden incurrir en las siguientes faltas: aplicar un volumen de muestra insuficiente; contaminar la muestra e insertar erróneamente el módulo de reactivos.
De igual forma, deben identificarse los peligros del mal uso razonablemente previsible. Por ejemplo, uso del DMD sin leer las instrucciones o completar la capacitación; ignorar las instrucciones u otra información de seguridad; usar un tipo de muestra o reactivo diferente al indicado por el fabricante; emplear los resultados para un uso clínico no indicado; las inadecuadas condiciones de almacenamiento de los materiales y reactivos; no realizar el mantenimiento prescrito de los instrumentos y reiterar el uso de materiales desechables.
Para cada peligro reconocido, se deben considerar las secuencias o combinaciones de eventos razonablemente previsibles que pueden provocarlo y deben identificarse y documentarse los daños resultantes. Una situación peligrosa es aquella circunstancia en la que los pacientes están expuestos a uno o más riesgos.
Se deben estimar los riesgos asociados a cada situación, teniendo en cuenta la probabilidad de que ocurra y la gravedad del posible daño; esto requiere la comprensión del uso clínico de los resultados del examen de diagnóstico. También deben identificarse los perjuicios a los pacientes con especificidad para asignarles valores de gravedad adecuados. Si bien la gravedad es una magnitud continua, en la práctica se simplifica su análisis.6) A continuación se muestra cómo analizarla en cinco niveles:
Despreciable: resulta en molestia o incomodidad temporal.
Menor: resulta en herida o discapacidad temporal que no requiere intervención médica o quirúrgica.
Serio/mayor: resulta en herida o discapacidad que requiere intervención médica o quirúrgica.
Crítico: resulta en discapacidad permanente o herida irreversible.
Catastrófico/fatal: resulta en la muerte.
Para estimar la probabilidad de ocurrencia del daño, se necesita analizar las combinaciones de eventos razonablemente previsibles que conducen a una situación peligrosa. La descripción de la secuencia de eventos debe comenzar con un suceso (generalmente la causa del peligro que se analiza) y los eventos directamente bajo el control del fabricante, luego debe progresar a través de las acciones de los usuarios del dispositivo y terminar con las decisiones clínicas que convierten los daños identificados en resultados previsibles.
Una situación peligrosa inicial puede ser la falla del Sistema de gestión de la calidad (SGC) del fabricante en cualquier etapa del diseño, desarrollo, producción y posproducción del DMD. Están bajo el control del usuario, los errores de uso e insuficiencias del dispositivo durante su funcionamiento en el sitio de atención médica; para prevenirlos y detectarlos deben tenerse en cuenta las medidas de protección y la información de seguridad del fabricante. Los DMD pueden constituir un riesgo para los pacientes por el uso indebido, las fallas en los procedimientos de aseguramiento de la calidad, los planes de contingencias y la recuperación o protección de seguridad.
Bajo el control del médico están la recepción y revisión del examen, así como la decisión de aceptarlo como válido. Es probable que los resultados con parámetros elevados se cuestionen, y hasta se rechacen (por ser contradictorios o incompatibles con la vida), pero los resultados plausibles podrían aceptarse como válidos y utilizarse para decisiones médicas. El análisis de riesgos debe considerar cualquier uso clínico razonablemente previsible. El uso clínico contraindicado o no abordado en la documentación adjunta al DMD podría considerarse un uso indebido razonablemente previsible a los efectos de la gestión de riesgos. En la guía se dan ejemplos de decisiones y actividades que están bajo el control del médico con la orientación y el apoyo del laboratorio.8
La probabilidad de que un paciente sufra daños resulta de la combinación de cada evento en la secuencia de eventos asociados con un peligro particular y el daño potencial. Se debe reunir un equipo multifuncional de expertos que estime la probabilidad de perjuicios en cada situación peligrosa identificada y utilice como complemento la información clínica disponible y el desempeño del DMD.
Para secuencias complejas o combinaciones de eventos, segmentar la situación peligrosa permite un análisis más eficiente de los expertos. Esto se denomina enfoque “P1 x P2” porque la probabilidad final resulta de multiplicar la probabilidad P1 de que ocurra una situación peligrosa (relacionada con el análisis en el laboratorio que utiliza el DMD y produce el resultado) y la probabilidad P2 de que se produzca un daño como resultado de esa situación peligrosa (relacionada con el uso del resultado por parte del médico y las decisiones y acciones basadas en ese resultado); ambas probabilidades se estiman por separado por los expertos correspondientes.
En la guía se dan preguntas para estimar la probabilidad de la situación peligrosa en el contexto del laboratorio y se ejemplifican los aspectos que ayudan a estimar la posibilidad de inconvenientes en el entorno clínico.8 El análisis de los expertos puede ofrecer estimaciones cualitativas o semicuantitativas de que una situación peligrosa genere un daño. Las estimaciones cualitativas de la probabilidad pueden ser alta (es probable que pase siempre o frecuentemente); media (puede suceder, pero no frecuentemente) y baja (ocurre remotamente durante el tiempo de vida del DMD).
La valoración de riesgos es el proceso de comparación del riesgo estimado frente a los criterios de riesgo establecidos por el fabricante en su plan de gestión para determinar su aceptabilidad. Cuando se realiza la estimación de la gravedad y la probabilidad de riesgos, se puede construir una matriz de doble entrada en la que se representa el riesgo aceptable y no aceptable y se colocan los riesgos estimados (tablas 1 y 2).
Tabla 1 Matriz con tres niveles de probabilidad y gravedad
| Alta | R2 | |||
| Media | R1 | R4, R6 | R5 | |
| Baja | R3 | |||
Leyenda: Riesgo aceptable: R1, R3 Riesgo inaceptable: R2, R4, R6, R5
Fuente: Normas de referencia
Tabla 2 Matriz con cinco niveles de probabilidad y gravedad
| R2 | ||||||
| R3 | ||||||
| R6 | R1 | |||||
| R4 | ||||||
| R5 | ||||||
Leyenda: Riesgo despreciable: R5 Riesgo de mayor análisis: R4 Riesgo inaceptable: R1, R2, R3, R6
Fuente: Normas de referencia
En la forma numérica se asignan valores a los diferentes niveles cualitativos y se calcula un número de prioridad del riesgo (NPR) por el producto de los valores asignados a la gravedad (S) y la probabilidad (P):
Cuando se emplea la expresión numérica para valorar el riesgo se puede incluir la detectabilidad (D) y se utiliza la fórmula siguiente:
Se muestra a continuación un ejemplo de los niveles de detectabilidad del peligro:
Alta: los controles establecidos permiten una detección eficaz de la falla. Casi siempre es detectable.
Media: comportamiento moderado de los controles de detección. Hay ocasiones en que la falla no se detecta.
Baja: los controles no son capaces de detectar las fallas, nunca o casi nunca.
Cuando se valora el riesgo por el NPR y este tiene un valor por encima de lo considerado no aceptable, el fabricante debe realizar actividades de control de riesgos: proceso en el que se toman decisiones y se implementan medidas para mantener el riesgo dentro de los niveles especificados. Estas medidas deben centrarse en reducir la probabilidad de los eventos peligrosos bajo el control del fabricante. Además, debe verificarse la efectividad de la información para la seguridad en el laboratorio.
Las acciones de control de riesgos van encaminadas al diseño y la fabricación seguros. Los estándares internacionales para DMD abordan aspectos de la seguridad inherente, en correspondencia con al estado de la técnica. Las medidas de protección en el DMD o en el proceso de fabricación se relacionan con los sistemas de detección de fallos, controles de la calidad en proceso y final; inspecciones en la recepción de materias primas y los componentes suministrados; alarmas y mensajes de error para alertar a los usuarios sobre condiciones de falla y procedimientos de recuperación.
Se debe proporcionar información de seguridad a los usuarios de DMD para prevenir una situación peligrosa. La norma ISO 18113-2:20098 ofrece la información de seguridad para los DMD in vitro y para los DMD complejos, se brindan programas de capacitación para evitar errores de uso. El fabricante debe implementar las medidas de control de riesgos y debe verificar su eficacia. Esta comprobación se puede realizar como parte de la validación del diseño y desarrollo del equipo y puede incluir ensayos con usuarios. También puede realizarse en la calificación del proceso dentro de un SGC.
Después de la implementación de las medidas de control, el fabricante debe evaluar el riesgo residual a partir de los criterios de aceptabilidad definidos en el plan de gestión de riesgos. Cuando el riesgo residual no se considera aceptable, se deben considerar otras medidas de control. Si se determina que la reducción del riesgo no es factible, se debe analizar la relación perjuicio-beneficio. El DMD se admite cuando los riesgos son menores que los beneficios. Se debe informar a los usuarios sobre los riesgos residuales para que puedan sopesarlos a la hora de utilizar el DMD y tomar decisiones informadas sobre la aceptabilidad del riesgo.
El productor también debe revisar los efectos de las medidas de control de riesgos si aparecen situaciones peligrosas nuevas o si los riesgos estimados se ven afectados por las medidas de control de riesgo. Todo riesgo nuevo o incrementado se debe gestionar de acuerdo con las orientaciones ya descritas. Asimismo, se debe revisar que se hayan considerado las situaciones peligrosas identificadas y se hayan completado las actividades de control de riesgos.
Después de implementar y verificar las medidas de control, se debe evaluar el riesgo residual global del DMD. Para ello, se deben considerar todos los riesgos residuales en relación con los beneficios del uso previsto a partir del método y los criterios de aceptabilidad definidos en el plan de gestión de riesgos. Si el riesgo residual global se considera aceptable, se debe informar a los usuarios sobre los más significativos e incluir la información necesaria en la documentación que acompaña al producto. En cambio, si se considera inaceptable en relación con los beneficios, se pueden implementar medidas adicionales, modificar el DMD o su uso previsto; de no ser así, el riesgo residual global permanece inaceptable.
El fabricante debe establecer un sistema eficaz para el seguimiento de la información de posproducción (quejas, eventos adversos y no conformidades del producto). Además debe monitorear los peligros y sus causas a partir de la retroalimentación con los usuarios que experimentan los eventos. Los informes de fallas de dispositivos, errores de uso e incidentes médicos deben recopilarse y analizarse. Hay que comparar las frecuencias observadas con las previstas para evaluar si los laboratorios notifican los eventos.
Discusión
Los dispositivos médicos de diagnóstico poseen un conjunto de especificidades, incluidos sus requisitos metrológicos, que se tienen que considerar para un adecuado proceso de gestión de riesgos. Además de las características biológicas, químicas, eléctricas, mecánicas y de seguridad que son comunes con otros equipos médicos, los DMD deben tener un desempeño y confiabilidad característicos que determinen la idoneidad para su uso clínico. El incumplimiento de un requisito de desempeño, confiabilidad o comunicación puede iniciar una secuencia de eventos dañinos para el paciente.
La guía se basa en el estudio de documentos internacionales, que aún no están disponibles como normas cubanas, pero pueden ser de utilidad tanto para los fabricantes, como para los formadores, consultores y evaluadores.
Un elemento medular en la gestión de riesgos es el análisis del riesgo-beneficio que debe tener en cuenta la capacidad de un DMD para identificar una enfermedad específica, proporcionar un diagnóstico en diferentes etapas de la enfermedad, predecir una enfermedad o identificar a los pacientes que responden a una terapia determinada. Debe realizarse el seguimiento del desempeño clínico para conocer la gravedad de los daños y su frecuencia, como fuente de retroalimentación para la gestión del riesgo.
La decisión de adoptar una nueva tecnología médica requiere de un análisis exhaustivo de la relación entre los beneficios y los riesgos asociados al proceder clínico y a las circunstancias de uso del producto. Por consiguiente, la guía para la gestión de riesgos de los DMD representa una herramienta útil para los diseñadores, fabricantes, evaluadores, personal médico y asesores.

