










 Serviços customizados
Serviços customizados Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroducción
La enfermedad de Alzheimer (EA) es considerada la demencia más común (60 al 70 %), y se encuentra entre las seis afecciones valorada por la Organización Mundial de la Salud (OMS) como una prioridad en relación con la salud mental.1 Se calcula que a escala mundial, 25 millones de personas padecen esta enfermedad, con un pronóstico de 75 millones para el 2030, y 131 millones en 2050. Dado que el número de pacientes con EA aumentará en el futuro, la carga económica y social que representan estos pacientes será enorme.
Cuba es un país en vías de desarrollo, con indicadores de salud similares a los de países desarrollados, y con un rápido envejecimiento de su población total; el 20,1 % de sus ciudadanos son mayores de 60 años. Actualmente es el país de Latinoamérica y del Caribe con mayor proporción de adultos mayores. De acuerdo con investigaciones realizadas sobre la prevalencia de la enfermedad, se estima una cantidad de 150 000 personas con la EA u otro tipo de demencia, y para el año 2030 se espera alcance la cifra de 273 000. Sin una intervención efectiva; es decir, si no se logra una cura para esta enfermedad en los próximos años, el número de cubanos con demencia se incrementará en 2,3 veces para el 2040 (300 000 personas con demencia), lo que significa el 2,7 % de la población cubana.2
Una de las características neuropatológicas de la EA es la presencia de placas β-amiloides y ovillos neurofibrilares en el cerebro, los cuales se asocian con el proceso que conduce a la degeneración progresiva y a la muerte neuronal. Las placas se localizan en el exterior de las células y están formadas, fundamentalmente, por depósitos de péptidos β-amiloides de 42 aminoácidos (βA1-42). La estructura predominante de las proteínas amiloidogénicas es la lámina β-cruzada.3,4 Varios autores han demostrado que la región comprendida entre los aminoácidos 8 al 22 se comporta como la más amiloidogénica y es la responsable de la formación del cierre de la estructura β-plegada.5,6
Otra característica importante de la EA es la hiperfosforilación de la proteína Tau. Esta es una proteína asociada a microtúbulos que se encuentra principalmente en el axón de las neuronas maduras, y su disfunción causa la neurodegeneración y la demencia en muchos pacientes.7,8
Existe una diversidad de hipótesis sobre la patogenia de la EA. Sin embargo, las de mayor aceptación son la “Hipótesis de la Cascada del Amiloide” y la “Hipótesis Colinérgica”. La primera, se basa en la acumulación aberrante de péptidos amiloideos que comienzan a agregarse y a formar estructuras tóxicas (placas seniles) en el cerebro. Esto inicia una cascada de respuestas metabólicas a nivel celular que ocasiona alteraciones en la homeostasia neuronal, disfunciones sinápticas, la muerte celular, y finalmente la demencia. Por otro lado, la Hipótesis Colinérgica se basa en el déficit de la acetilcolina (ACh), neuro-transmisor encargado de la transmisión del impulso nervioso, que provoca un desbalance neuronal, y origina un deterioro del funcionamiento cognitivo.9,10
Existen potenciales blancos terapéuticos para retardar o inhibir el proceso de aparición de la EA, entre ellos, la inhibición de la agregación y la producción del péptido βA1-42 y la inhibición de la enzima acetilcolinesterasa (AChE).
El péptido βA1-42 con conformación β, está compuesto por 42 aminoácidos, y permanece en equilibrio con los oligómeros y las protofibrillas difusibles circundantes. Los agregados de βA1-42 son estructuras que aparecen de forma temprana, antes de que los síntomas de la enfermedad se establezcan.11) La enzima AChE, presenta una cavidad de 20 Å de profundidad en donde se han identificado dos zonas fundamentales para la actividad biológica. La primera es el sitio aniónico periférico PAS (por sus siglas en inglés Positive AnionicSite) formado por los aminoácidos (a.a) Trp 84, Trp 279 y Phe 330. La segunda es la tríada catalítica, y está formada por los a.aSer 200, Glu 327 e His 440, y se denomina sitio aniónico catalítico CAS (Catalitic Anionic Subsite).12
Varias moléculas se han diseñado con el objetivo de lograr la desagregación y eliminación de las placas formadas por el péptido βA1-42, y de restablecer el equilibrio celular; y con ello, el funcionamiento cognitivo (ejemplo Tramiposate, Solanezumab, Gantenerumab y Aducanumab.13 El uso de los fármacos antinflamatorios no esteroideos también ha demostrado su efectividad contra esta enfermedad (ejemplo Naproxeno e Ibuprofeno).14
En el caso de los inhibidores específicos para la AChE, el uso de Rivastigmina, Donepezilo, Galantamina y derivados de Tacrina ha logrado mejorar las disfunciones cognitivas en los pacientes que sufren la EA en etapas moderada y leve. Sin embargo, su uso debe constreñirse a un período de 12 a 18 meses; a partir del cual los pacientes empiezan a empeorar cognitivamente.15
Desde el pasado siglo se han evaluado, a través de técnicas computacionales, diferentes compuestos capaces de interactuar con el péptido βA1-42, con las enzimas AChE y ϒ y β-secretasas, y con la proteína Tau, entre otros blancos terapéuticos. Los métodos más utilizados para ello son el desarrollo de modelos de relación estructura actividad (QSAR, por sus siglas en inglés Quantitative Sructure-Activity Relation ship) y las simulaciones de acoplamiento molecular. Todas estas investigaciones se dirigen a tratar de seleccionar compuestos líderes, sobre la base de la afinidad hacia el blanco, el tipo de interacción, la estabilidad conformacional y energética, entre otros criterios.16
El objetivo del presente trabajo es obtener dos modelos QSAR para la predicción de la actividad inhibitoria de dos series de compuestos diferentes sobre dos blancos relacionados con la EA; así como determinar, a través de las técnicas de acoplamiento molecular, el sitio, el tipo de interacción y la estabilidad conformacional que se establece entre los compuestos evaluados y los blancos seleccionados.
A partir de ellos se predice para las dos series de compuestos seleccionados, la actividad inhibitoria de la agregación del péptido βA1-42 y de la enzima acetilcolinesterasa (AChE). Ambas series están constituidas por moléculas que contienen anillos aromáticos como motivo estructural central. La combinación de los dos modelos QSAR obtenidos y los estudios de acoplamiento molecular, permitirá la identificación y selección de los mejores compuestos como posibles líderes contra los blancos terapéuticos.
Materiales y métodos
Un descriptor molecular se define como el resultado final de un procedimiento lógico y matemático que transforma la información procedente de ciertas propiedades de las moléculas (tanto teóricas como experimentales) que es codificada dentro de una representación simbólica en un número útil para predecir, mediante correlaciones estadísticas, el resultado de un determinado experimento o propiedad medible. Las “familias” de moléculas generalmente se seleccionan a partir de semejanzas estructurales, y se definen sustituyentes o fragmentos moleculares en diferentes “posiciones” que las distinguen entre sí. El conjunto de factores que permite explicar la actividad de una familia de moléculas puede ser parametrizado para los diferentes sustituyentes que la constituyen. De este modo, se han ideado descriptores de los efectos de dichos sustituyentes sobre la capacidad de una molécula determinada para interaccionar con su receptor.16
Por todo lo anterior, en los estudios QSAR, la actividad biológica se representa en función de estos descriptores como se muestra a continuación:

Cálculo de los diferentes descriptores moleculares
Se empleó el visualizador molecular Avogadro 17) para modelar las moléculas de los ligandos. El programa Gaussian se empleó para optimizar dichas geometrías al nivel de teoría B3LYP/6-31G(d) 18,19), donde se calcularon las frecuencias vibracionales para verificar si se ha alcanzado un mínimo de la superficie de energía potencial. Los descriptores electrónicos para el estudio QSAR se extraen de las geometrías optimizadas. Otros descriptores: log P (coeficiente de partición octanol/agua), energía de hidratación (EH2O), polarizabilidad (P), área superficial aproximada (ASA), volumen molar (VM) y la refractividad molar (RM); se realizaron con el programa HYPERCHEM 7, a partir de las geometrías optimizadas anteriormente.20)
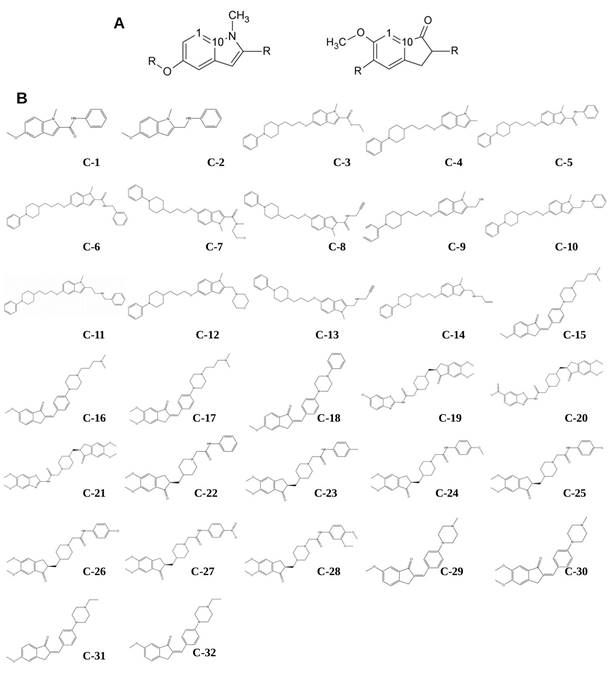
Las estructuras de las moléculas utilizadas en las bases de datos de los inhibidores del péptido-βA y de los inhibidores de la acetilcolinesterasa se muestran en las figuras 1 y 2, respectivamente.21,25
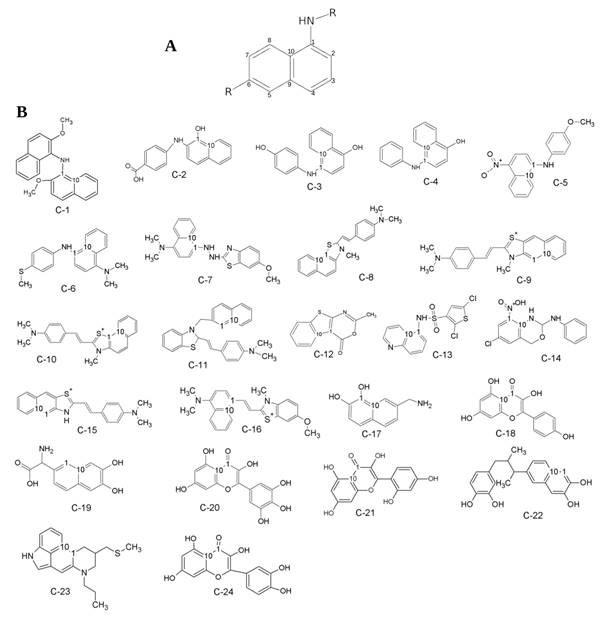
Fig. 1 Estructuras químicas de los 24 compuestos utilizados en la confección del modelo QSAR para la predicción de la actividad antiagregante del péptido βA1-42 (Serie 1). [21-23] A) Farmacóforo; B) Compuestos de la Serie 1
Análisis estadístico de los datos
Modelos de regresión lineal múltiple
La selección de variables en los modelos de regresión lineal múltiple (RLM) se realizó con la aplicación del algoritmo genético (AG), implementado en el programa Build QSAR. En este programa, el AG tiene la siguiente configuración: 1) operadores genéticos: de mutación, cruzamiento y selección; 2) función de evaluación: el coeficiente de correlación (R); 3) número de variables: 3 o 4, según el modelo utilizado; 4) tamaño de la población: 100; 5) número de generaciones: 200; 6) por ciento mutación: 10.26)
El análisis de RLM fue empleado para obtener modelos específicos que relacionan las estructuras y las propiedades cuánticas y químico-físicas de las moléculas seleccionadas en las bases de datos. El análisis estadístico se desarrolló con el programa STATISTICA 7.27)
La búsqueda del mejor modelo QSAR fue procesada en términos del mayor valor de coeficiente de correlación (R), el valor de la prueba de F: Fisher, el p-valor (p < 0,01) y el menor valor de desviación estándar (s). La calidad de los modelos fue determinada al examinar estos parámetros estadísticos y los procedimientos de validación cruzada.27)
Análisis de conglomerados
El análisis de conglomerados (AC) se realizó para demostrar la diversidad estructural de la base de datos utilizada en el estudio QSAR. Se realizó un AC jerárquico implementado en el programa STATISTICA 7.27) En este estudio, se emplea el enlace completo (complete linkage) como el método para fusionar las moléculas en los conglomerados (cluster). Para calcular la distancia entre las moléculas, se utilizó la distancia euclidiana.28)
Preparación de las estructuras para las simulaciones ligando-proteína
En este estudio, se seleccionaron 55 compuestos divididos en dos series, 23 de ellos para la base de datos de los compuestos inhibidores de la agregación βA1-42 (Serie 1, el compuesto 17 se excluyó por ser un outlayer) 21,23), y los restantes 32 para los inhibidores de la enzima AChE (Serie 2).24,25 Las coordenadas tridimensionales (3D) se generaron a través de una optimización preliminar en Avogadro 1.1.1 17), con el empleo del algoritmo de pasos descendentes y el campo de fuerza de mecánica molecular MMFF94 (por sus siglas en inglés Molecular Mechanic Force Field).29)
La estructura 3D del péptido amiloideo 1iyt fue seleccionada del PDB.(11) Este fichero presenta una estructura obtenida por técnicas de RMN con diez conformaciones distintas del péptido βA1-42 en disolución. Para separar cada una de las diez configuraciones, se utilizó el programa VMD (Visual Molecular Dynamics).30) La estructura 3D de la enzima AChE fue resuelta mediante difracción de rayos X (código de PDB 1eve, con resolución de 2,5 Å) [12], y se obtuvo de la base de datos del PDB.
A partir de las estructuras en formato PDB del péptido βA1-42, de la enzima AChE, de los ligandos, y con el auxilio del programa AutoDock Tools, se obtuvieron los ficheros de entrada en formato “.pdbqt”, los que se construyen para llevar a cabo las simulaciones de acoplamiento molecular a través del programa Autodock Vina (VINA).31)
Para realizar el acoplamiento molecular en la estructura del péptido, se mantienen rígidos todos los enlaces de las cadenas laterales de los aminoácidos.
Identificación de la zona de interacción ligando-proteína
Preparación de los ficheros de configuración
Todas las simulaciones de acoplamiento molecular ligando-proteína se realizaron con el programa VINA.(31) En el caso de los ligandos, las cargas parciales se calcularon mediante el modelo de Gasteiger. [30] Los átomos de hidrógeno no polares se fusionaron a los átomos pesados. Se mantuvieron los enlaces con libre rotación por defecto, con el auxilio de la herramienta TORSDOF del Auto Dock Tools.32) Para las proteínas seleccionadas, las cadenas laterales de todos los residuos se consideraron rígidas en un primer estudio. Para realizar el anclaje molecular de los complejos ligando-blanco, se construyó una caja de simulación que abarcó a toda la proteína. El centro de la caja de simulación se define en un punto, en donde se hizo “coincidir” con el origen del eje de coordenadas, y se utilizó una exhaustividad de 16.
El cómputo se llevó a cabo con el empleo de ficheros de configuración, así como con los ficheros individuales de las estructuras de los ligandos y del blanco. Los ficheros de configuración se desarrollaron con el lenguaje de programación Python, 33) y se obtuvieron múltiples ficheros, uno por cada una de las simulaciones deseadas. Cada fichero utilizó, preferencialmente, los parámetros del VINA. Para la selección de estos datos se utilizó el subprograma Autodock/Vina Plugins incluido en Pymol.
Análisis de las interacciones ligando-proteína
Los diferentes confórmeros de cada complejo, obtenidos de estas simulaciones, fueron sometidos a un proceso de agrupamiento con el empleo de un RMSD menor a 2 Å (Root Mean Square Deviation), como criterio de la distancia entre cada uno de los resultados. De esta forma, se clasificaron de acuerdo con el sitio y la forma en la cual interaccionan los ligandos con la proteína.34)
Para seleccionar los a.a que interaccionan con cada compuesto, se utilizó como distancia de corte 4,6 Å. Todos los contactos en el acoplamiento molecular que se repiten en el 85 % o más de todas las soluciones obtenidas, están incluidos en la zona de interacción entre los ligandos y la proteína en cuestión.
Resultados y discusión
Selección de las series de entrenamiento y de prueba usando análisis de conglomerados
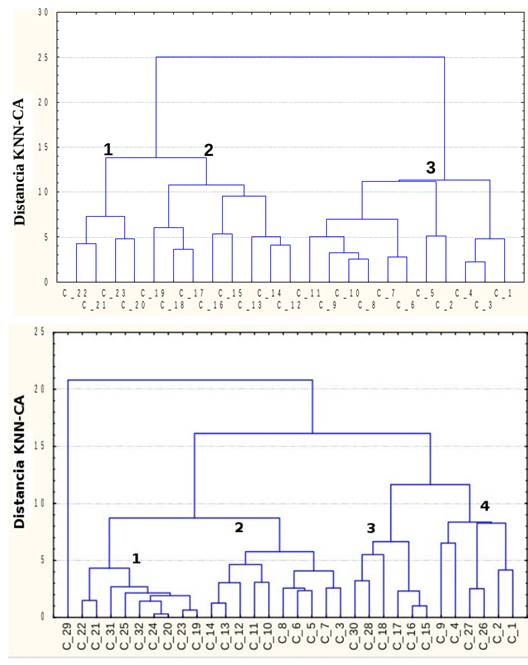
Como resultado del análisis de conglomerado, se agruparon las moléculas de acuerdo con sus semejanzas, y de cada conglomerado se extrajeron aleatoriamente compuestos para formar dos series: una de entrenamiento y la otra de prueba. Con este método se garantiza que en ambas series haya una representación de todas las moléculas de los conglomerados.
En la figura 3 (A y B) se observan los dendogramas correspondientes a los compuestos inhibidores de la agregación del péptido βA1-42 y a los inhibidores de la acción de la enzima AChE, respectivamente. Dentro de un mismo conglomerado, los compuestos presentan mayores semejanzas en cuanto a estructura química, tipo de enlaces, distancias interatómicas, entre otras características. En la figura 3A se observa, por ejemplo, que los compuestos C-15 y C-13 presentan semejanza estructural entre ellos, por lo que se agrupan en el conglomerado número 2.
Modelo QSAR para los inhibidores de la formación de agregados moleculares de βA1-42
Las variables seleccionadas por el algoritmo genético, incluidas en el modelo QSAR de la ecuación (3.1) fueron: la energía del orbital ocupado de mayor energía (HOMO, del inglés Highest Occupied Molecular Orbital) y las cargas parciales sobre los átomos de carbono 2 (q2) y de carbono 1 (q1) de la estructura aromática (Serie 1, ecuación 3.1).21,23)
Del análisis por conglomerados se seleccionaron las moléculas: C-5, C-6, C-11, C-16 y C-20 para serie de prueba. Con esta serie es posible comparar la predicción externa correspondiente a la ecuación (3.1)

Al analizar la calidad estadística del modelo se observa que el coeficiente de determinación (R2) obtenido en la serie de entrenamiento fue de 0,87 para los valores experimentales del logaritmo negativo del valor de la concentración inhibitoria 50 (pIC50). El modelo tiene un valor de coeficiente de determinación del procedimiento de validación cruzada (q2) de 0,79. Este valor de q2 > 0,5 puede ser considerado una prueba de la alta habilidad predictiva del modelo que se corrobora con la buena predicción de la serie de prueba externa (R2).35-36)
Modelo QSAR para los inhibidores de la acción de la enzima AChE
El modelo QSAR incluyó las variables siguientes: coeficiente de reparto octanol-agua (Log P), carga parcial sobre el átomo de carbono 1 (q1) de la estructura aromática, entalpía asociada al proceso (ΔH) y cantidad de elementos aceptores de enlaces de hidrógeno (acrónimo:naccr). Estas se muestran en la ecuación (3.2) junto con los estadígrafos más relevantes (Serie 2, ecuación 3.2). (24,25
Del análisis por conglomerados se seleccionaron las moléculas: C-1, C4, C-8, C-12, C-15 y C-24 para serie de prueba. Con esta serie fue posible comparar la predicción externa correspondiente a la ecuación (3.2)

Similar al caso anterior, se observa que la calidad estadística del modelo R2 fue de 0,83 para los valores experimentales de pIC50. El modelo tiene un valor de coeficiente de determinación del procedimiento de validación cruzada (q2) de 0,64, lo que indica la alta habilidad predictiva del modelo que se corrobora con la buena predicción de la serie de prueba externa (R2).
Los descriptores moleculares obtenidos para la ecuación 3.1 muestran que la dependencia del IC50 se favorece si disminuye el valor de la energía del orbital HOMO (descriptor electrónico). Esto se corresponde con la densidad de carga presente en el sistema, dada las nubes electrónicas π pertenecientes al núcleo aromático.
En la ecuación 3.1, el aumento de las cargas parciales q1 y q2 sobre las posiciones 1 y 2 de los carbonos en los núcleos bases de las moléculas respectivamente, siempre presenta una contribución negativa. En el caso de la ecuación 3.2, solamente depende la actividad inhibitoria de la carga sobre el átomo de carbono1 (q1). Esto sugiere que la presencia de grupos electrodonores, enlazado directa o indirectamente a estos átomos, podrían incrementar la densidad de carga negativa y con ello la actividad biológica predicha al contribuir a la estabilización del sistema ligando-diana. Estos resultados concuerdan con los obtenidos en la literatura para sistemas moleculares con similitudes a los estudiados.37,38)
Por último, en el caso de la ecuación 3.2, el modelo muestra que existe dependencia con la entalpía y con la cantidad de aceptores de enlaces de H en la molécula. La disminución del valor de la entalpía implica que el sistema es más estable al evolucionar a un mínimo de energía, y por tanto, mejor será el compuesto. En cuanto a la cantidad de grupos aceptores, mientras mayor sea su número (por debajo de diez, según el criterio establecido por Lipinski en 1997) 39), más favorable será la interacción del compuesto con la AChE. Esto pudiera favorecer su interacción con los a.a de la región del PAS, y así tener una orientación correcta hacia los a.a del CAS.
Con el objetivo de validar la confiabilidad del modelo obtenido, se calcularon los valores de pIC50 para cada compuesto de ambas bases de datos (tablas 1 y 2).
Tabla 1 Valores de pIC50 calculados y pIC50 experimentales para la base de datos de compuestos con actividad antiagregante del péptido βA1-42 (Serie 1). [21-23] Descriptores obtenidos para la ecuación (3.1)
| Código | HOMO | q1 | q2 | pIC50calc | pIC50exp |
|---|---|---|---|---|---|
| C-1 | -0,18 | 0,16 | 0,11 | 5,31 | 5,24 |
| C-2 | -0,20 | 0,18 | 0,13 | 5,48 | 5,59 |
| C-3 | -0,17 | -0,21 | -0,21 | 5,91 | 6,00 |
| C-4 | -0,18 | 0,27 | 0,11 | 5,19 | 5,27 |
| C-5 | -0,20 | 0,33 | 0,06 | 5,44 | 5,57 |
| C-6 | -0,18 | 0,29 | 0,07 | 5,23 | 5,66 |
| C-7 | -0,19 | 0,07 | 0,08 | 5,59 | 5,60 |
| C-8 | -0,30 | -0,19 | 0,09 | 7,11 | 6,68 |
| C-9 | -0,30 | -0,25 | 0,15 | 7,13 | 6,92 |
| C-10 | -0,29 | -0,28 | 0,12 | 7,14 | 6,68 |
| C-11 | -0,29 | -0,24 | 0,14 | 7,06 | 6,92 |
| C-12 | -0,22 | 0,32 | 0,19 | 5,58 | 5,68 |
| C-13 | -0,20 | -0,10 | 0,16 | 5,85 | 5,92 |
| C-14 | -0,22 | 0,34 | -0,12 | 5,86 | 5,80 |
| C-15 | -0,30 | -0,23 | 0,13 | 7,09 | 6,80 |
| C-16 | -0,29 | 0,31 | 0,09 | 6,47 | 6,37 |
| C-18 | -0,21 | 0,32 | -0,02 | 5,60 | 5,49 |
| C-19 | -0,21 | -0,24 | -0,71 | 6,82 | 5,77 |
| C-20 | -0,21 | 0,42 | -0,68 | 6,08 | 6,29 |
| C-21 | -0,20 | 0,32 | -0,02 | 5,53 | 6,17 |
| C-22 | -0,20 | -0,26 | 0,17 | 5,96 | 5,96 |
| C-23 | -0,19 | 0,15 | -0,01 | 5,59 | 5,70 |
| C-24 | -0,21 | 0,32 | -0,02 | 5,59 | 6,14 |
Tabla 2 Valores de pIC50 calculados y de pIC50 experimentales para la base de datos de compuestos con actividad inhibitoria de la enzima acetilcolinesterasa (Serie 2). [24-25] Descriptores obtenidos para la ecuación (3.2)
| / |
Log P | q1 | Δ |
naccrb | pIC50 cal | pIC50exp |
|---|---|---|---|---|---|---|
| C-1 | 6,63 | 0,38 | -1,52 | 3 | 4,91 | 4,00 |
| C-2 | 7,36 | 0,38 | -1,41 | 3 | 5,97 | 4,82 |
| C-3 | 7,82 | 0,37 | -2,21 | 5 | 6,89 | 6,57 |
| C-4 | 7,66 | 0,38 | -2,08 | 4 | 5,82 | 6,40 |
| C-5 | 6,09 | 0,38 | -2,48 | 4 | 3,23 | 4,00 |
| C-6 | 7,35 | 0,38 | -2,42 | 4 | 4,70 | 4,00 |
| C-7 | 6,26 | -0,51 | -2,83 | 3 | 6,90 | 6,57 |
| C-8 | 7,07 | 0,38 | -2,17 | 5 | 6,17 | 6,89 |
| C-9 | 7,96 | 0,37 | -1,96 | 4 | 6,45 | 6,62 |
| C-10 | 7,34 | 0,38 | -2,24 | 5 | 6,29 | 6,85 |
| C-11 | 7.60 | 0,38 | -2,36 | 5 | 6,26 | 7,08 |
| C-12 | 7,24 | 0,38 | -2,24 | 5 | 6,19 | 7,00 |
| C-13 | 6,81 | 0,38 | -2,05 | 4 | 5,01 | 6,72 |
| C-14 | 6,81 | 0,38 | -2,05 | 5 | 6,18 | 6,85 |
| C-15 | 5,57 | 0,13 | -2,11 | 4 | 5,11 | 5,20 |
| C-16 | 6,53 | 0,16 | -2,11 | 4 | 5,90 | 5,07 |
| C-17 | 6,9 | 0,13 | -2,29 | 4 | 6,05 | 5,22 |
| C-18 | 6,79 | 0,13 | -2,08 | 3 | 5,29 | 5,31 |
| C-19 | 7,19 | 0,38 | -2,11 | 4 | 5,22 | 6,51 |
| C-20 | 7,58 | 0,38 | -2,18 | 4 | 5,52 | 6,38 |
| C-21 | 6,54 | 0,38 | -2,24 | 5 | 5,43 | 6,46 |
| C-22 | 6,59 | 0,37 | -2,30 | 5 | 5,37 | 6,59 |
| C-23 | 5,5 | 0,38 | -2,11 | 4 | 3,45 | 4,00 |
| C-24 | 7,31 | 0,37 | -2,18 | 4 | 5,26 | 4,74 |
| C-25 | 5,55 | 0,37 | -2,20 | 5 | 4,53 | 4,00 |
| C-26 | 6,81 | 0,32 | -1,93 | 5 | 6,82 | 7,24 |
| C-27 | 5,65 | -0,18 | -1,75 | 3 | 6,81 | 7,37 |
| C-28 | 7,24 | 0,34 | -2,02 | 5 | 6,94 | 7,14 |
| C-29 | 6,43 | 0,39 | -2,81 | 6 | 5,07 | 10,06 |
| C-30 | 6,79 | 0,34 | -1,95 | 5 | 6,66 | 8,74 |
| C-31 | 5,65 | -0,20 | -2,11 | 4 | 7,22 | 6,72 |
| C-32 | 5,20 | -0,20 | -2,18 | 4 | 6,60 | 6,46 |
aΔH: entalpía; bnaccr: cantidad de elementos aceptores de enlaces de hidrógeno
En la figura 4 se correlacionan los valores de pIC50 experimentales y los calculados para las dos series de compuestos. Como se observa, existe una linealidad entre los valores (R2 = 0,87 y R2 = 0,85, respectivamente), que indica que los cálculos teóricos son similares a los experimentales. Esto sugiere que los modelos son confiables bajo las condiciones y los descriptores utilizados para cada uno de los casos.
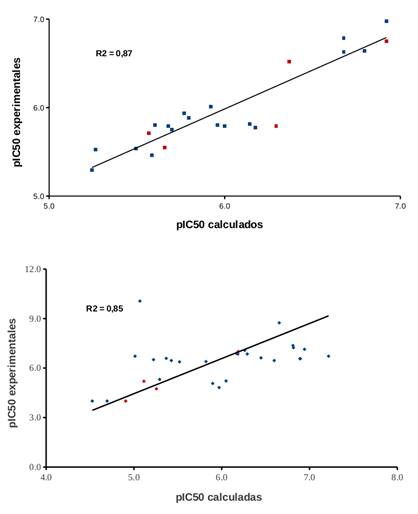
Fig. 4 Valores de pIC50 calculados vs pIC50 experimentales. A) Compuestos de la Serie 1, ecuación (3.1); B) Compuestos de la Serie 2, ecuación (3.2)
Para obtener información de la región teórica del espacio químico, definida por los descriptores del modelo, se determinó el dominio de aplicabilidad (DA). Este es “el rango dentro del cual el modelo tolera una nueva molécula”, y se define como un área dentro de los límites +2 y -2 para los residuales estandarizados y un umbral del valor de influencia para la actividad inhibitoria.
En este estudio, los compuestos con residuales estandarizados superiores a dos desviaciones estándares se consideran no confiables.40) Las predicciones son poco fiables para los compuestos con un valor de influencia mayor que el valor crítico (h = 3(p + 1)/n), donde p es el número de variables del modelo y n es el número de compuestos (figura 5).
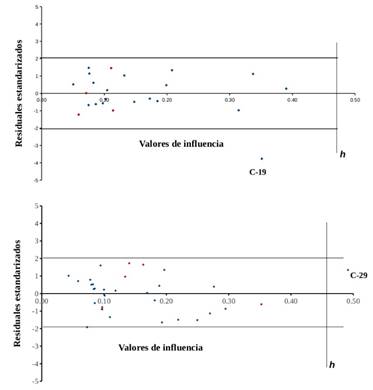
Fig. 5 DA de cada modelo obtenido. Azul: conjunto de entrenamiento; rojo: conjunto de prueba. A) DA de la Serie 1, ecuación (3.1), h = 0,48; B) de la Serie 2, ecuación (3.2), h = 0,47
El parámetro p toma valores de 3 para el modelo de la ecuación (3.1), y de 4 para la ecuación (3.2). El valor crítico h es de 0,48 para la ecuación (3.1), y de 0,47 para la ecuación (3.2). Como se observa en la figura 5, en ambos modelos existen compuestos que tienen un valor de influencia superior al valor crítico; por tanto, la actividad de estos compuestos sobre las dianas terapéuticas no puede explicarse por las ecuaciones obtenidas en este estudio.
Estudios de acoplamiento molecular ligando-blanco terapéutico
En la identificación de la posible zona de interacción entre cada uno de los compuestos y los blancos βA1-42 (Serie 1) y AChE (Serie 2), se realizaron cien simulaciones de acoplamiento molecular a cada ligando con la estructura de cada proteína (código de PDB: 1iyt y 1eve, respectivamente). Así, se obtuvieron alrededor de 5 500 estructuras del complejo ligando-proteína.
En el proceso de conglomerado del total de las simulaciones se obtuvo que, en la mayoría de los casos, más del 90 % de todos los compuestos presenta una superposición estructural elevada, con un valor de RMSD de 2 Å. El grupo con mayor número de soluciones y menor energía promedio, se seleccionó como el modo de unión representativo para cada complejo, y se utilizó para los cálculos y las representaciones.
Estudios de acoplamiento molecular con el péptido βA1-42
En la tabla 3 se observan los a.a del péptido βA1-42 involucrados en la interacción con cada uno de los 23 compuestos seleccionados en este estudio. Estos están comprendidos en la región amiloidogénica del a.a 8 al 22.5-6)
Tabla 3 Aminoácidos de la región amiloidogénica (a.a 8 al 22) del péptido βA1-42 que interactúan con cada uno de los ligandos de la Serie 1. Se incluye el por ciento de contactos para cada compuesto por la zona seleccionada
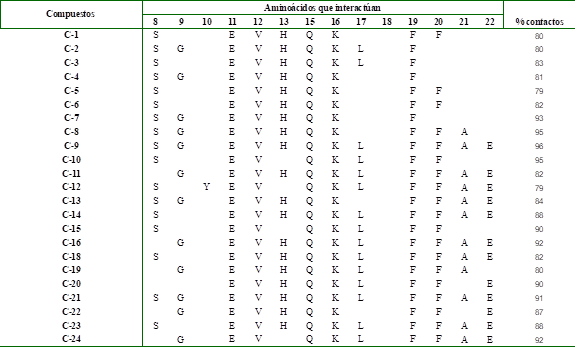
*Código de una letra de aminoácidos
De ellos, 18 interaccionan con la Ser 8, aminoácido polar poco iónico que forma enlace de H con los grupos -NH2, -NH, -OH y -COOH presentes en los compuestos. Existen 13 compuestos que presentan interacciones hidrofóbicas con la Gly 9 y 15 con la Leu 17, a través de los grupos fenilo y naftilo presentes en sus estructuras. Asimismo, dos compuestos presentan interacciones polares con la Tyr 10 y los grupos -OH y -COOH (compuestos C-27 y C-31, respectivamente). Un total de veinte compuestos presenta interacciones con la His, trece mediante la formación de enlaces de H, entre los grupos -OH, -NH y el átomo de oxígeno del carboxilo de los compuestos.
La mayoría de los compuestos establecen interacciones polares con el Glu 11 y Glu 22, con la Gln 15 y con la Lys 16, preferentemente con los grupos -OH, -NH, -NH2 y con el carboxilo perteneciente a los ligandos de la base de datos. Asimismo, todos presentan interacciones hidrofóbicas entre las nubes electrónicas π de sus anillos aromáticos y los aminoácidos Val 12 y Phe 19 y Phe 20.
Todos los ligandos interactúan con el péptido βA1-42 a una distancia no mayor de 4,6 Å, principalmente con los aminoácidos Glu 11, Val 12, His 13, Gln 15, Lys 16, Leu 17, Phe 19, Phe 20, Ala 21 y Glu 22.
De acuerdo con los valores de IC50 calculados con la ecuación (3.1), según se muestra en la tabla 1 para cada compuesto, se selecciona como ejemplo dos ligandos distintos para ilustrar las interacciones que se establecen entre ellos y la diana seleccionada. Los compuestos seleccionados fueron los que presentaron mayor (C-7) y menor (C-19) correlación entre los valores pIC50 calculados y experimentales (figura 6). En líneas discontinuas se observan los enlaces de H formados en cada uno de los complejos.
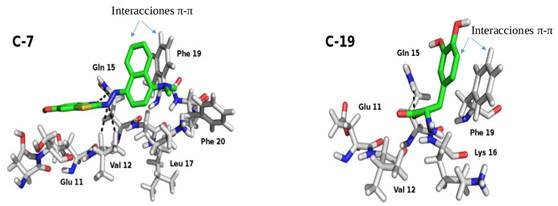
Fig. 6 Interacciones de los compuestos que presentan mayor y menor diferencia entre los valores de IC50, respectivamente (C-7 y C-19) y el péptido βA1-42, (PDB 1iyt)
En el compuesto C-7, las interacciones tienen lugar principalmente a través de enlaces de H e interacciones hidrofóbicas. Se observaron enlaces de H entre los átomos de N de la cadena amidoalquílica con los a.a Gln 15 y Val 12 del péptido βA1-42, lo que probablemente aumente la estabilidad del complejo ligando-péptido formado.
Las interacciones hidrófobas, a través de la nube π-π, se localizaron entre el grupo aromático de los ligandos y los grupos fenilo de los residuos a.a. Phe 19 o Phe 20. En general, estos resultados concuerdan con las predicciones obtenidas por Sablón-Carrazana, 41) en donde se utilizó el modelo de fibrillas de la secuencia del péptido βA17-42.
Para el compuesto C-19, la fortaleza de las interacciones disminuye, presumiblemente debido a la presencia de un solo anillo aromático en su estructura. Es de notar, que la longitud de la cadena carbonada es muy pequeña, lo que imposibilita el acoplamiento con la zona más amiloidogénica del péptido. Sin embargo, la presencia de enlaces de H con el residuo aminoacídico de Gln 15, le confiere cierta estabilidad.
De manera general, aunque la diferencia en la formación del complejo es apreciable, en ambos casos existen dos tipos de interacciones que evidencian la capacidad que tienen estos 35 ligandos de acoplarse al péptido βA1-42. Primero, las interacciones hidrofóbicas (π-π) que se establecen entre los grupos aromáticos de los ligandos con los residuos a.a Phe 19 o Phe 20, y segundo, las interacciones polares (principalmente enlaces de H) con los residuos Gln 15 y Val 12. Los resultados obtenidos sugieren que es posible que los compuestos puedan bloquear o inhibir el proceso de agregación del péptido al interactuar por la misma zona, e impedir la formación de los agregados.
A partir de estas simulaciones de acoplamiento molecular es posible obtener una función de puntuación, como medida de la afinidad del complejo formado. Para ello, se lleva a cabo un tamizaje virtual, con la base de datos por la zona de interacción seleccionada, mediante el programa VINA. El resultado del cálculo de la función de puntuación para el naproxeno, como molécula de referencia, fue de -4,3 kcal/mol. A este valor, se adiciona la desviación estándar para estos cálculos (± 2,85 kcal/mol) y se obtiene un valor de corte inferior de -7,15 kcal/mol.31) La mayoría de los valores de energía de los complejos formados están comprendidos entre el valor de corte inferior y el valor del naproxeno (tabla 4).
Como puede apreciarse en la tabla 4, ningún compuesto presenta diferencias con respecto al valor del naproxeno. Sin embargo, en el compuesto C-7 (-6,2 kcal/mol) presenta el mayor valor de la función de puntuación, lo que coincide con los resultados mostrados en la tabla 1, como uno de los que mejor predice el modelo QSAR. Asimismo, los valores de los compuestos C-17 y C-19 son los más bajos. Esto concuerda con los resultados mostrados en la figura 5A, donde se observa que ambos ligandos están fuera del DA para la ecuación (3.1) del modelo QSAR.

Estudios de acoplamiento molecular con la enzima AChE
En la tabla 5 se observan los resultados de las simulaciones de acoplamiento de los compuestos con actividad inhibitoria sobre la enzima AChE. Se representan solamente las interacciones que se establecen con los a.a tanto del PAS como del CAS.
Tabla 5 Interacción de cada compuesto de la Serie 2 con los a.a del PAS y del CAS de la enzima acetilcolinesterasa, con más de un 90 % de contactos
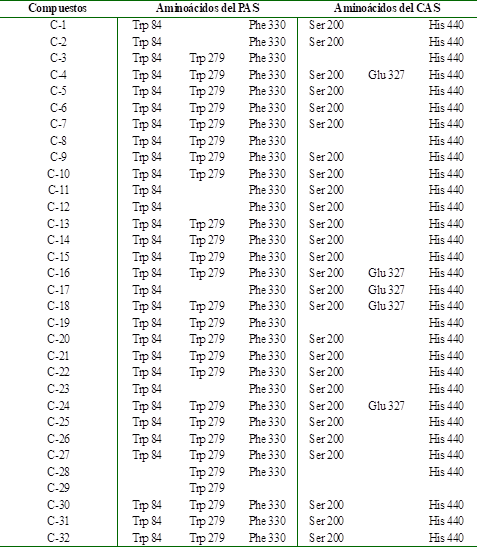
La mayoría de estos ligandos presentan interacciones con los residuos de Ser 200 e His 440, los cuales forman parte del sitio activo de la enzima. Ellos influyen en la atracción electrostática del sustrato hacia el sitio donde se producirá la hidrólisis (característica de las enzimas del grupo serino-hidrolasas como la AChE). [15] De los 32 compuestos, 24 presentan interacciones hidrofóbicas con todos los residuos de la región del PAS (Trp 84, Trp 279 y Phe 330). Este tipo de asociación permite orientar, de manera adecuada, a los compuestos en la región catalítica de la enzima.
En la figura 7 se muestran las interacciones entre los compuestos C-18 y C-29, tomados como ejemplos de este estudio con comportamientos diferentes, y la enzima acetilcolinesterasa
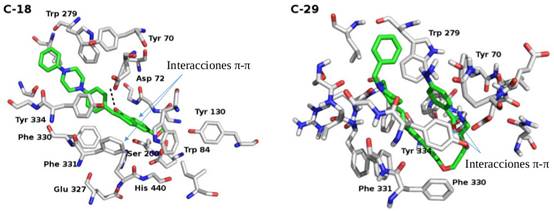
Nótese en la figura, la interacción con los a.a del PAS (Trp 84, Trp 279 y Phe 330) y del CAS (His 440, Glu 327 y Ser 200) para el caso del complejo C-18-AChE. Como se observa, el compuesto C-18 presenta interacciones con todos los a.a pertenecientes tanto a la región del CAS como del PAS. Las interacciones entre los anillos aromáticos de los compuestos y los residuos de fenilalanina y triptófano permiten que se “anclen” en el bolsillo aromático formado por los a.a Phe 288, Phe 290, Phe 330, Phe 331, Trp 279 y Trp 84. Este sitio se encuentra en el interior de la cavidad en donde se produce la acción enzimática y bloquea el posible paso de otros compuestos. Además, se observan interacciones polares del tipo enlace de H con el residuo aminoacídico Asp 72, lo cual le confiere estabilidad al complejo formado.
En cambio, el compuesto C-29 no tiene interacciones con los residuos de la cavidad, excepto con Phe 330, Phe 331 y Trp 279. Tampoco se producen interacciones polares con ningún otro aminoácido. Esto pudiera ocurrir debido a que este ligando se localiza hacia la parte más expuesta al disolvente, y no hacia el interior de la cavidad de interacción, en donde se encuentran los residuos de Ser 200, His 440 y Glu 327, pertenecientes a la tríada catalítica de la región del CAS. Sin embargo, en la mayoría de los ligandos evaluados se observa interacciones polares, principalmente enlaces de H, que comprenden a los átomos donores-aceptores presentes en la cadena carbonada de los compuestos. Estas interacciones se establecen específicamente con los a.a Asp 72, Trp 84, Gly 117, Gly 118, Tyr 121 y Tyr 130.
Los resultados del tamizaje virtual por la zona de interacción seleccionada, con el empleo del programa VINA se muestran en la tabla 6.
Tabla 6 Valores del cálculo de la función de puntuación (afinidad) para los complejos ligando-AChE obtenidos mediante el programa Autodock Vina

Donepezilo: compuesto de referencia
Como se observa en la tabla 6, el valor de la función de puntuación en el caso del Donepezilo (referencia) fue de -9,1 kcal/mol. A este valor se adiciona la desviación estándar para este procedimiento (±2,85 kcal/mol), y así se obtiene el valor de corte inferior (-11,95 kcal/mol). La mayoría de los complejos formados presentan valores comprendidos en el intervalo entre el corte inferior y el calculado para el Donepezilo, lo cual puede deberse a la presencia de enlaces de H.
De los compuestos estudiados, C-15 y C-18 mostraron valores de la función de puntuación por encima del Donepezilo; -12,2 y -12,4 kcal/mol, respectivamente. Por el contrario, la molécula C-29 es el que menor valor presenta (-7,5 kcal/mol). Esto, en conjunción con el análisis del tipo de interacciones de este compuesto con el sitio catalítico de la enzima, sugiere que no debe actuar como un inhibidor de la acción enzimática. Este resultado concuerda con lo obtenido en el estudio del DA dado por la ecuación (3.2), donde el compuesto C-29 queda fuera de los compuestos que el modelo es capaz de predecir.
Conclusiones
El modelo QSAR propuesto para cada serie permite describir la contribución los descriptores moleculares a la actividad biológica. El primer modelo describió la dependencia de la actividad biológica del péptido βA1-42 en correlación con la energía del orbital HOMO y las cargas parciales de los átomos de carbono C1 y C2. El segundo modelo describió la dependencia del IC50 de los ligandos con la enzima AChE en correlación con el Log P, la carga parcial sobre el átomo de carbono C1, la entalpía y la cantidad de aceptores de hidrógeno en la molécula. Ambos modelos son significativos y predictivos, de acuerdo con los valores obtenidos para los estadígrafos R2 y q2, respectivamente.
De acuerdo con el estudio de acoplamiento molecular, la mayoría de las interacciones relevantes observadas para el péptido βA1-42 y para la enzima la AChE son interacciones π-π. Estas se establecen entre los anillos aromáticos de los ligandos y los grupos aromáticos de los a.a Phe 19 o Phe 20 del péptido βA1-42y Phe 330, Phe 331, Trp 84 y Trp 279 de la enzima.
A través de este estudio se confirma, que la presencia de anillos aromáticos en los ligandos son la unidad central de la estructura de los compuestos biológicamente activos. En el caso específico de los enlaces de H, se formaron para el péptido βA1-42 entre los aminoácidos Glu11, His 13, Gln 15 y Lys 16 y para la enzima AChE entre los a.a Asp 72, Gly 117, Gly 118, Tyr 121, Tyr 130 y los átomos donores-aceptores presentes en la cadena carbonada de los compuestos de ambas series. Estas interacciones permiten orientar, de manera adecuada, a los compuestos en la mejor conformación del péptido y de la región catalítica de la enzima.
A partir de los modelos obtenidos para cada blanco terapéutico, es posible predecir los valores de IC50 de nuevos ligandos, que presenten semejanzas estructurales a los utilizados en cada una de las series estudiadas y seleccionar los compuestos candidatos a fármacos.

