










 Serviços customizados
Serviços customizados Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
El síndrome de Holt-Oram es un trastorno del desarrollo determinado genéticamente, con un patrón de herencia autosómica dominante, con alta penetrancia y expresividad muy variable; sin embargo, se han documentado varios casos como mutaciones de novo, es decir, nuevas. Esta enfermedad se presenta con gran heterogeneidad clínica en su expresión fenotípica, lo que dificulta la identificación de formas clínicas con variantes más ligeras. Se caracteriza clínicamente por una asociación de anomalías esqueléticas en las extremidades superiores de forma variable y asimétrica, con anomalías cardiovasculares. Tiene una incidencia estimada de 1/100 000 nacidos vivos.1,2
En este síndrome las anomalías esqueléticas afectan a las extremidades superiores y a la cintura escapular con una gran diversidad que oscila desde focomelia hasta mínima limitación de movimiento de los pulgares, los codos y los hombros. Las alteraciones de los pulgares son las más frecuentes y consisten en hipoplasia de la eminencia tenar, falta de oposición del pulgar, sindactília parcial, pulgares trifalángicos/digitalizados e hipoplasia o ausencia de pulgares. Otras anomalías esqueléticas que se han descrito son la hipoplasia de radios, las anomalías claviculares, la estrechez de la cintura escapular o la hipoplasia de la musculatura.2
En el aparato cardiovascular, entre las alteraciones más frecuentes, se describen la comunicación interauricular del tipo ostium secundum, la comunicación interventricular, el tronco arterioso o el canal auriculoventricular común. Como anomalías cardíacas menores se han descrito las alteraciones electrocardiográficas sin alteración estructural, las anomalías de la válvula mitral, las miocardiopatías o el examen cardiológico normal.3
Para realizar el diagnóstico clínico de este síndrome deben estar presentes las alteraciones características de la extremidad superior y la afectación cardíaca en el mismo individuo; pueden aparecer en sus progenitores defectos de grado variable y mostrarse evidencia de la transmisión genética, de lo contrario puede plantearse como una nueva mutación. Por lo general no existe correlación entre la gravedad de las anomalías esqueléticas y las cardíacas.4,5
En 1997 fue descubierto el gen responsable, llamado TBX5, que ha sido mapeado y clonado en 12q24.1. Es la primera localización cromosómica de un gen responsable de defectos congénitos del septo cardíaco en seres humanos. El TBX5 pertenece a una familia de genes (T-box transcription factor family) implicados en el desarrollo y la diferenciación del mesodermo y debido a que el comienzo del desarrollo cardíaco y de las extremidades superiores tienen lugar entre la tercera y la cuarta semanas de vida embrionaria.6
Este paciente, además de la alteración en la extremidad superior izquierda y la comunicación interauricular, nació con otro defecto congénito, una atresia esofágica que ha sido la causa de varias operaciones. Por lo particular del caso se considera interesante su descripción, que enfatiza la expresividad variable de esta enfermedad; es necesario tenerla presente para poder realizar el diagnóstico clínico.
INFORMACIÓN DEL PACIENTE
Paciente masculino que nació a las 37,2 semanas, parto eutócico por inducción por disminución del líquido amniótico, con peso de 2 980 gramos, talla de 51 cm y una prueba de Apgar de 7-9; de inmediato los Especialistas en Neonatología le diagnosticaron una atresia esofágica y fue enviado al Centro Provincial de Genética del Hospital Pediátrico Universitario “Octavio de la Concepción de la Pedraja”, hospital de referencia de la Cirugía neonatal, de la Cuidad de Holguín, de la provincia del mismo nombre; cuatro horas después fue intervenido quirúrgicamente y se le realizó una gastrostomía que hasta hoy mantiene. El tramo de atresia esofágica fue extenso, con bordes muy distantes, y requiere, para su reposición normal, una operación compleja, con injerto de intestino, que no se ha podido efectuar aún. Posteriormente, en la Sala de Cirugía, fue examinado nuevamente y se observó que el brazo izquierdo era más pequeño en la porción mesomélica (corresponde a ulna y radio) y se le auscultó un soplo de grado II-III/VI; se le indicó un ecocardiograma que mostró una comunicación interauricular en forma de ostium secundum. Se mantuvo hemodinámicamente estable, sin tratamiento, hasta que cerró espontáneamente alrededor del primer año.
Estuvo desnutrido los seis primeros meses de vida y delgado hasta los tres años, con retardo en el desarrollo motor que logró mejorar al corregirse su estado nutricional; consiguió caminar, sin apoyo, a los tres años, con buen desarrollo del lenguaje.
Actualmente, con cuatro años, tiene buen desarrollo pondoestatural y mantiene entre el 10 y el 25 percentil, con desarrollo del lenguaje acorde con su edad.
En el examen físico se valoró el acortamiento del brazo izquierdo a expensas de la región radial, con hipoplasia de este, al igual que el primer artejo de la mano izquierda, que también tiene implantación alta (Figura 1).
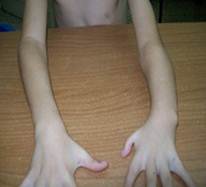
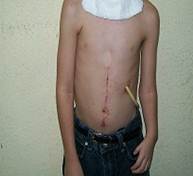
El defecto de los miembros es asimétrico, como se observa en la Figura 2, con acortamiento del miembro superior izquierdo, producto de la hipoplasia radial. En esta misma figura se puede valorar la extensa cicatriz en la región abdominal producto de las operaciones para corregir el defecto congénito, atresia esofágica, que aún no se ha logrado y fue necesario realizar una gastrostomía que aún mantiene.
Por lo poco habitual de encontrarse este síndrome asociado a malformación digestiva, se le indicaron exámenes para efectuar diagnóstico diferencial con otras patologías.
Realizándose radiografías cervicales, torácicas y lumbares, que fueron normales y ultrasonido abdominal descartándose otras malformaciones. Se realizó toma de muestra de sangre periférica para cariotipo convencional a 400 bandas que observaron 18 metafases, resultando un varón cromosómicamente normal 46, XY.
Se les solicitó a la familia consentimiento informado para examinarlo y realizar todos los demás estudios, así como para tomarle fotografías y publicar en revistas científicas, a lo cual accedieron sus padres.
Después de estos estudios por el patrón dismórficos y malformativo se discute en colectivo y se interconsulta con especialistas de mayor experiencia y se llega al diagnóstico clínico de síndrome Holt-Oram.
DISCUSIÓN
Los síndromes cardiomiélicos abarcan la cardiopatía congénita y las malformaciones esqueléticas de los miembros superiores y están relacionados con mutaciones en los factores de transcripción con los dominios de T-Box. Produciéndose probablemente una alteración de empalme del gen y resultando en una proteína truncada en su extremo C-terminal. El síndrome Holt-Oram es causado por una mutación dominante en el gen TBX5 que altera la estructura tridimensional de la proteína y su función de unión al ADN. Encontrándose varias mutaciones puntuales y deleciones en TBX5 en estos pacientes.1,2,3
Los padres de este afectado no presentan ninguna alteración, por lo que se planteó como una nueva mutación, aunque no se descarta un posible mosaicismo gonadal.3
Esta enfermedad con significativa heterogeneidad clínica pero también heterogeneidad genética obstaculiza el diagnóstico, ya que la localización y el tipo de mutación en TBX5 no son predictores de la expresividad fenotípica y la probabilidad de encontrar la mutación en el gen TBX5 es solo del 74%. Así varios estudios reportan mutaciones del tipo de duplicaciones, deleciones parciales en otros cromosomas como el 14, inversión pericéntrica del cromosoma 20.3,4
En 1997 se estudió y comprobó que la mutación del gene humano TBX5, es factor crítico para el desarrollo de extremidades y corazón, sugiriendo que la insuficiencia haploide del gene TBX5 es la etiología molecular de esta enfermedad. Demostrándose en 1999 que la mutación TBX5 causa anomalías substanciales en corazón y pulgares, observándose distintos fenotipos en el cambio de glicina por arginina (gly 80 arg), que causan malformaciones cardiacas graves, pero sólo mínimas alteraciones en el esqueleto; mientras que cuando ocurren dos mutaciones, arginina por glicina (237, arg237 a gly) y arginina por triptófano (arg a trp), existen extensas lesiones de extremidad superior, pero lesiones menos significativas en el corazón. Esto pudiera explicar en parte la variabilidad y la severidad de algunas presentaciones y esta última pudiera ser lo ocurrido en este caso.5,6
El diagnóstico diferencial debe realizarse con entidades como la asociación VACTERL, que exhibe malformaciones vertebrales, atresia anal, cardiopatías congénitas, fistula traqueo esofágicas, estenosis esofágicas, así como malformaciones renales, con displasia de las extremidades, donde son de gran importancia las alteraciones en vertebras, que este apaciente no tiene. La pancitopenia de Fanconi, que tiene alteraciones de miembros y las alteraciones hematológicas que no presenta este caso. Con el síndrome Poland, que presenta agenesia de pectoral mayor de un hemicuerpo, pudiendo tener afectación de grado variable del miembro superior del mismo lado. Otro diagnóstico diferencial se realiza con la trombocitopenia-ausencia de radio, que no tiene este caso y la embriopatía por talidomida, que se descarta por no existencia en nuestro medio y encontrarse retirado desde los años setenta a nivel mundial porque se comprobó su efecto teratógeno. Otros investigadores han reportado casos asociados con atresia aortica, malformación anorectal, también asociación con microcefalia, opacidad del cristalino.7,8
Sin embargo las malformaciones renales y digestivas no son tan comunes, así como la atresia esofágica como presenta este paciente, solo otro anteriormente reportado además de la malformación renal también presentó atresia gastroesofágica que fue causa de intervención quirúrgica de urgencia.9
Esta enfermedad exhibe heterogeneidad clínica y genética con amplia variabilidad fenotípica, que pudiera tener diagnóstico prenatal ultrasonográfico, en los casos con expresión severa del defecto en miembros y en aquellos en los que se busca por existencia de otro afectados en la familia. Se puede encontrar como un hallazgo al asociarse en el ultrasonido genético defecto cardiovascular con malformación de miembros superiores que hace pensar en este síndrome.10
En Cuba no contamos con diagnóstico molecular para este síndrome malformativo por lo que resulta de vital importancia estudiar cada caso en particular, así como sus familiares utilizando el método de clínico o de patrón para delinear bien el fenotipo. Llegar al diagnóstico clínico permite tomar conductas oportunas e informar a las familias de la posible morbilidad y mortalidad de estos pacientes, dependiendo de la gravedad de la cardiopatía congénita, que en su mayoría requieren intervención de urgencia. Considerándose necesario realizar un adecuado asesoramiento genético a cada familia afectada, explicando el riesgo de recurrencia para la descendencia que es alto del 50 %.

