










 Serviços customizados
Serviços customizados Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroducción
La genética médica en el mundo ubica sus primeros pasos entre 1865, fecha en que Mendel publicó su trabajo.1 En 1953 aparece el modelo molecular del ADN (ácido desoxirribonucleico) propuesto por Watson y Crick.2
Las enfermedades genéticas que afectan la piel y sus anexos representan del 15-20 % de los casos dada la frecuencia de expresión de las mismas.3 La Organización mundial de la salud (OMS) reconoce a Cuba como un país que incorporó los servicios de genética médica en los tres niveles de atención del sistema nacional de salud.4 En Las Tunas las genodermatosis representan 22,22 %.5 Las más frecuentemente diagnosticadas dentro de los trastornos de la pigmentación fueron las mastocitosis, con 11,66 % y las neurofibromatosis, con 8,33 %.5,6
Debido a la importancia de conocer estas enfermedades, se decidió realizar este trabajo con el objetivo de compilar información actualizada acerca de las características y criterios diagnósticos de enfermedades genéticas que faciliten su estudio y el seguimiento de los pacientes.
Métodos
Se realizó la revisión de la literatura disponible en SciELO, PubMed Central, Medline Plus, Clinicalkey, Orphanet y OMIM. Los descriptores utilizados fueron genética médica, enfermedades dermatológicas genéticas. Durante el proceso de revisión, se consultaron 87 bibliografías entre libros y artículos publicados en las bases de datos, de los cuales 22 fueron citados dentro de las referencias bibliográficas, de ellos 7 libros y 16 artículos publicados en los últimos 5 años, con 78,26 % de actualización.
Análisis y síntesis de la información
Las genodermatosis son enfermedades de origen genético cuya expresión fenotípica en la piel constituye su manifestación clínica principal o diagnóstica. Constituyen un grupo de afecciones heterogéneas7,8 que tienen en común, mecanismos vinculados con los genes (Cuadro 1).
Cuadro 1 Genodermatosis y principales características genéticas
| Desorden | Herencia | Locus del gen |
|---|---|---|
| Incontinencia pigmenti | XD | Xq28 |
| Lentiginosis familiar generalizada | AD | 4q21.1-q22.3 |
| Síndrome de LEOPARD | AD | 12q22-qter |
| AD | 12q24 | |
| AD | 3p25 | |
| AD | 7q34 | |
| Mastocitosis | AD | 4q12 |
| Neurofibromatosis tipo I | AD | 17q11.2 |
| Síndrome de Watson; neurofibromatosis- síndrome de Noonan | AD | 17q11.2 |
| Neurofibromatosis tipo II | AD | 22q12.2 |
| Neurofibromatosis tipo VIII | AD | 12q13 |
| AD | 14q13 | |
| Síndrome de Noonan | AD | 15q22.31 |
| AD | 12p12.1 | |
| AD | 12q22 | |
| AD | 12q24.3 | |
| AD | 7q34 | |
| AD | 3p25 | |
| AD | 2p22-p21 | |
| AD | 1p13.2 | |
| AD | 1p15.2 | |
| AD | 1q22 |
Incontinencia pigmenti (ORPHA 308300)
También conocida como síndrome de Bloch-Sulzberger, es un complejo síndrome multisistémico en el cual las lesiones cutáneas se asocian con defectos en ojos, sistema musculoesquelético y sistema nervioso central. La incontinencia pigmenti se clasifica en 2 tipos, una forma familiar y una esporádica.9,10 Tiene un patrón de herencia dominante ligado al cromosoma X (Fig.1).
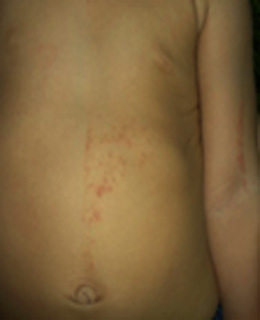
En 1906 Garrod describió el primer caso, en 1926 Bloch reportó un caso y propuso los posibles mecanismos para los cambios pigmentarios característicos. El nombre de incontinencia pigmenti surgió en este mismo período y Sulzberger, años más tarde, informa un caso con muchos detalles, citado por William y otros.9
Criterios diagnósticos
Criterio mayor
Las lesiones cutáneas siguen un patrón blaschkoide y pasan por cuatro fases: la primera se inicia durante la vida intrauterina y perdura hasta dos semanas después del nacimiento. Se caracteriza por brotes de pápulas eritematosas y vesículas de disposición lineal y localización acral. La segunda fase se extiende desde las dos semanas de vida hasta los pocos meses y se caracteriza por la aparición de pápulas verrucosas, desarrolladas sobre las lesiones vesiculosas previas. La tercera fase se produce entre el primer y cuarto mes de la vida y en ella aparecen unas máculas hiperpigmentadas lineales de color gris pizarra, que siguen las líneas de Blaschko, localizadas principalmente en el tronco y las extremidades y pueden aparecer tanto en las zonas afectadas como en las respetadas por la erupción inicial. La cuarta fase perdura durante varios años, se caracteriza por desaparición lenta de las máculas hiperpigmentadas y la aparición en su lugar de máculas hipopigmentadas con cierto grado de atrofia. Esas lesiones no se broncean, ni en ellas se produce sudoración. Además, existe aplasia cutis congénita (25 %) y cambios ungueales.
Criterios menores
Presentan alteraciones del sistema nervioso central (25 %): retardo mental, retardo en el desarrollo motor, paraplejia; alteraciones oculares (30 %); alteraciones dentales (50 %): dentición retardada, falta de dientes (colmillos superiores y premolares); alopecia; uñas anormales; alteraciones retinianas típicas; pelo lanoso; antecedentes de múltiples abortos; alteraciones esqueléticas.
Se diagnostica con el criterio mayor y uno o más menores.10,11
La biopsia de piel demuestra una dermatitis con vesículas subcórneas, con abundantes eosinófilos. El estado verrugoso se caracteriza por una hiperqueratosis y una inflamación crónica en la dermis. En la etapa pigmentaria la melanina se encuentra libre en la dermis, o englobada en los macrófagos dérmicos, con ausencia o disminución de la melanina en las células basales de la epidermis (esta característica es la que da el término de incontinencia pigmentí).10
Síndrome LEOPARD (OMIM 151100, ORPHA 500)
Es un trastorno genético multisistémico poco frecuente caracterizado por lentigos, miocardiopatía hipertrófica, talla baja, malformación torácica y rasgos faciales dismórficos. Se transmite en forma autosómica dominante, alta penetrancia y expresividad variable.12,13
Koller y otros, en su artículo “Síndrome de LEOPARD a propósito de la mancha café negro” exponen que la enfermedad se describe por primera vez en 1936 por Zeisler y Becker; y en 1969 Gorlin y otros, propusieron el acrónimo que resume las principales manifestaciones clínicas que dan nombre al síndrome:
L: Lentiginosis
E: Electrocardiograma (ECG) alterado
O: Hipertelorismo Ocular
P: Estenosis Pulmonar
A:Anomalías genitourinarias
R: Retraso del crecimiento
D: Deafness (sordera neurosensorial) (Fig.2).
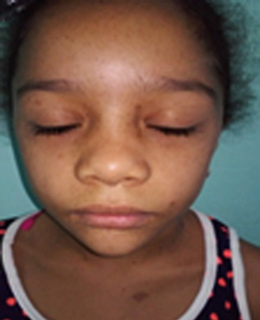
Manifestaciones clínicas
Las manifestaciones dermatológicas constituyen el primer signo visible de esta genodermatosis. Los lentigos son los elementos característicos. Están presentes desde el nacimiento o aparecen durante la niñez y son muy numerosos en la etapa adulta. Se localizan en piel y mucosas y afectan áreas expuestas y no expuestas, incluidas palmas y plantas.
En general respetan labios y mucosa oral. Consisten en máculas hiperpigmentadas, cuya coloración varía del pardo al pardo-negruzco, de 2-5 mm. de diámetro, de contornos netos, en ocasiones irregulares por coincidir con la desembocadura de los ostium foliculares. También pueden presentar máculas de varios centímetros de diámetro, generalmente congénitas, de coloración negruzca denominadas “manchas café negro” ya que tienen cierta similitud con las “manchas café con leche” pero de coloración más oscura.
Otras manifestaciones cutáneo-mucosas observadas son: pigmentación periorbitaria, hiperpigmentación de la línea media subumbilical, pliegue simiano bilateral, onicopatías, máculas hipopigmentadas, hiperelasticidad cutánea y esteatocistomas múltiples.
Presentan cambios electrocardiográficos, fundamentalmente bloqueo y hemibloqueo de rama y desviación del eje hacia la izquierda. Entre los defectos estructurales se destacan la estenosis pulmonar (40 %), estenosis mitral, estenosis valvular subaórtica, fibrosis endomiocárdica. También pueden presentar miocardiopatía hipertrófica obstructiva, signo infrecuente que constituye la principal causa de muerte del síndrome.
Dentro de las manifestaciones esqueléticas, Pectum excavatum y carinatum son las más frecuentes; se puede apreciar también, prognatismo mandibular, escoliosis, hiperlaxitud articular y escápula alata.
Entre las manifestaciones génitourinarias destaca la criptorquidea bilateral, hipospadias e hipoplasia genital. Las pacientes afectadas pueden presentar pubertad tardía y ovarios hipoplásicos. Las anomalías renales son infrecuentes, entre ellas, el riñón en herradura.
La sordera constituye el signo menos frecuente, es de carácter neurosensorial y en la mayoría de los casos se diagnostica al nacimiento o durante la primera infancia. Otros hallazgos vinculables al síndrome son retraso mental leve, electroencefalograma alterado e hipotonía muscular.
Generalmente presentan baja talla final con peso y talla normales al nacer.
El hipertelorismo ocular constituye el elemento hallado en 100 % de los casos, además pueden presentar fisuras o ptosis palpebral, narinas grandes e implantación baja de las orejas
El diagnóstico de la entidad es clínico: se basa en la presencia de lentiginosis y dos manifestaciones clínicas presentes en el acrónimo. En ausencia de lentiginosis puede diagnosticarse si presenta tres características del síndrome y un familiar en primer grado con diagnóstico de este síndrome de LEOPARD.12,13
Mastocitosis (OMIM 154800)
Comprende un espectro de enfermedades caracterizadas por un cúmulo de mastocitos en la piel que se traducen en la aparición de máculas hiperpigmentadas, con o sin afectación de otros órganos o sistemas. Al examen dermatológico el signo patonogmónico es el signo de Darier, que consiste en la formación de una roncha si se frota la lesión macular.14,15 (Fig.3).
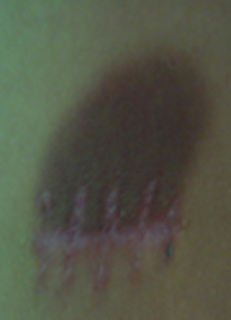
Las diferentes formas clínicas se clasifican según La Organización Mundial de la Salud (OMS) en:
Un mastocitoma solitario puede ocurrir en la niñez con una tendencia a la involución espontáneo. La urticaria pigmentosa es esporádica, caracterizada por macules, pápulas, o nódulos, raramente asociado con la enfermedad sistémica y típicamente aclara en la pubertad; en contraste, la urticaria pigmentosa del adulto persiste y puede asociarse con mastocitosis sistémica, sobre todo con afectación de la médula del hueso.14,15
La neurofibromatosis (OMIM 162200)
Se define como un trastorno neurocutáneo, de origen genético, caracterizado fundamentalmente por alteraciones melánicas de la piel y la presencia de tumores, como resultado de la afectación del tejido nervioso periférico, por lo que muy pocas estructuras quedan exentas de manifestaciones clínicas. (Cuadro 2),
La enfermedad se describió desde los inicios del siglo xiii y fue von Recklinghausen en 1882, quien introdujo el término “neurofibroma”, detalló las lesiones de piel y precisó el carácter hereditario de la enfermedad de ahí que sea conocida como enfermedad de von Recklinghausen, citado por Arenas y otros.16
Cuadro 2 Clasificación de las neurofibromatosis (clasificación de Ricciardi, 1982)
| Tipo | Variante | Rasgos clínicos característicos | Rasgos genéticos |
|---|---|---|---|
| I |
Formación clásica o enfermedad de von Recklinghausen |
Presencia de 6 o más manchas café con leche, nódulos de Lisch, múltiples neurofibromas. Pecas axilares. | Gen localizado cerca del centrómero del cromosoma 17. |
| II | Forma acústica | Neurinoma acústico bilateral. Pocas manchas café con leche. Menos neurofibromas cutáneos, tumores células Schwann de los nervios craneales o las raíces de los nervios, meningiomas, gliomas, cataratas subcapsular, ausencia de nódulos de Lisch, | Gen mapeado en cromosoma 22. |
| III | Forma mixta | Manchas café con leche que son pálidas, pocas en número y grandes. Neurofibromas cutáneos especialmente en las palmas. Múltiples tumores en el cerebro (neuromas acústicos, meningiomas, espinal/paraespinal neurofibromas) de comienzo temprano con curso rápido. Ausencia de gliomas ópticos y nódulos de Lisch. | Hasta el momento todos los casos son esporádicos. |
| IV | Forma variante | Presencia de manchas café con leche diseminadas, ausencia de neuro-fibromas o neurofibromas difusos, sin deformidades | - |
| V | Forma segmentaria |
No familiar. Diferenciación característica de las manchas café con leche o pecas axilares son ipsilaterales en áreas restringidas del cuerpo. Los neurofibromas no cruzan la línea media. |
Surge por una mutación somática postzigótica y generalmente no es heredable |
| V I | Síndrome de las manchas café con leche | Solo manchas café con leche. Rasgos no específicos, |
Debe darse en dos generaciones para poder ser diagnosticada |
| VII | Inicio tardío | Inicio después de los 20 años de edad. Neurofibromas no claros hasta el final de la tercera década o más tarde. Ausencia de manchas café con leche y nódulos de Lisch. | Todos los casos son esporádicos. |
| VIII |
No especificada o independiente |
Neurofibromatosis sin características de ninguna otra categoría. Neuro-fibromas se limitan al tracto gastrointestinal. |
El gen podría posiblemente estar unido a uno de los puntos de ruptura en las bandas de los cromosomas 12q13 y 14q13. |
| IX | Neurofirnomatosis tipo Noonan |
Combinación de rasgos de a neurofibromatosis y síndrome de Noonan. |
La mayoría de los casos son esporádicos hasta la fecha. Casos de familias en ocasiones. |
Los criterios diagnósticos de la neurofibromatosis tipo 1 (2 o más criterios) son:
Seis o más manchas café con leche de 5 mm en pacientes prepuberales y mayores de 15 mm en pospúberes.
Dos o más neurofibromas o un neurofibroma plexiforme.
Signo de Crowe (efélides axilares).
Glioma del nervio óptico.
Dos o más nódulos de Lish.
Lesiones óseas típicas (displasia de las alas esfenoidales o adelgazamiento cortical de huesos largos con o sin seudoartrosis.
Antecedentes de neurofibromatosis tipo I en padres o hermanos.16,17
Los criterios del 4 al 6 requieren de exámenes especializados para su diagnóstico como la tomografía o resonancia magnética nuclear (RMN), la visualización de los hamartomas retinianos a través de la lámpara de hendidura y la radiografía del lugar de la lesión, respectivamente. El resto de los criterios son clínicos.16
Los criterios diagnósticos de la neurofibromatosis tipo 2 son:
Masas nerviosas bilaterales en el octavo nervio que se detectan con tomografía o RMN.
Un pariente en primer grado con NF2 y también:
Se debe cumplir una de estas posibilidades.
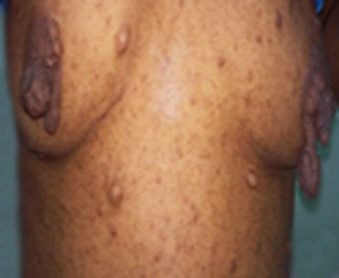
Se muestran neurofibromas y manchas café con leche en la neurofibromatosis tipo I (Figs. 4 y 5).
Síndrome de Noonan (ORPHA 648, OMIM 163950)
El Síndrome de Noonan fue descrito por Noonan y Ehmkeenen en 1963. La incidencia se ha estimado en 1 de 1000 y 1 de 2500 nacimientos vivos. Se hereda en forma autosómica dominante y tiene una expresividad muy variable.19,20,21
Clínicamente, se caracteriza por: hipocrecimiento de inicio posnatal precoz, fenotipo facial característico, defectos cardíacos (especialmente, estenosis pulmonar), un variable déficit cognitivo, cubitus valgus, deformidades torácicas y vertebrales, retardo mental, leves alteraciones hematológicas, retraso puberal y criptorquidia en varones, queratosis folicular predominantemente en superficie extensora y región facial, queratosis pilar atrófica facial, que puede conducir a la desaparición de las cejas, manchas café con leche y lentigos.19,20,21
La facie con cara triangular, frente amplia; hipertelorismo, fisuras palpebrales antimongoloides, implantación baja de la línea posterior del cabello, orejas rotadas de implantación baja y rotación hacia atrás con hélix grueso; anomalías oculares dadas por ptosis palpebral, hendiduras palpebrales antimongoloides, epicantus, estrabismo, proptosis, miopía y nistagmus; anomalías en el cuello que es corto o palmeado; maloclusión dentaria y micrognatia.19,20,21
Se muestran las características del síndrome de Noonan (Cuadro 3).
Cuadro 3 Características y criterios del síndrome de Noonan
| Criterios mayores | Criterios menores | Criterios diagnósticos |
|---|---|---|
| Típica | Sugerente | Facies |
| Estenosis valvular pulmonar, cardiomiopatía hipertrófica obstructiva o EKG típico | Otros defectos cardíacos | Cardíacos |
| < Percentil 3 | < Percentil 10 | Talla |
| Tórax ancho | Pared del tórax | |
| Familiar de primer grado con diagnóstico establecido de síndrome de Noonan | Pariente de primer grado con diagnóstico sugerente de síndrome de Noonan | Historia familiar |
| Presencia de las 3 anomalías | Presencia de alguna de las 3 anomalías |
Otros: Retraso mental Criptorquidia Displasia linfática |
Se establece el diagnóstico de síndrome de Noonan cuando:
Manchas gemelas o didimosis
La presencia concurrente, en una misma región, de dos áreas de tejido mutante, diferentes entre sí y del tejido normal circundante, es un fenómeno conocido como “manchas gemelas” o didimosis. Pueden deberse a mutaciones que suceden en el mismo alelo (didimosis alélica) o en distintos alelos (didimosis no alélica), son dos manchas diferentes que coexisten en el mismo individuo. Ejemplo de ello es la asociación de un nevo anémico con un nevo pigmentario y verrugoso.22)
Conclusiones
Se realizó una revisión bibliográfica en la cual se pudo apreciar que son múltiples las genodermatosis que cursan con trastornos de hiperpigmentación. Por tratarse de enfermedades en las que existe una alteración genética en que la expresividad varía de un individuo a otro, es necesario agrupar sus características y reunir sus criterios diagnósticos. Se presentaron además las didimosis, que por sus características pueden coexistir en ellas alteraciones hiperpigmentarias asociadas a hipopigmentación. Conocer qué las caracteriza, cómo diferenciarlas entre ellas y cómo realizar el diagnóstico favorecerá una mejor atención y educación de los pacientes para evitar complicaciones y mejorar su calidad de vida.

