










 Serviços customizados
Serviços customizados Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroducción
El síndrome de linfohistiocitosis hemofagocítica (LHH) es una enfermedad inmunomediada, potencialmente mortal. Fue inicialmente descrita por los pediatras Scott y Robb-Smith. Es causada por un compromiso en la función de las células T asesinas naturales y citotóxicas. Se trata de una entidad rara, con una incidencia anual acumulada de uno por cada 100 000 adultos.1,2,3
Existe poca evidencia sobre la LHH del adulto, especialmente en lo que respecta a la aproximación diagnóstica y terapéutica.1,4 La evidencia actual descansa fundamentalmente en series de casos y estudios no controlados y, por tanto, las decisiones diagnósticas y terapéuticas continúan basándose en la escasa experiencia clínica y en el criterio de expertos.1,5
Cuba no escapa a este fenómeno, existen solo siete informes de casos con este síndrome publicados hasta la fecha.6,7,8,9,10,11,12 Esta situación hace que cobren validez todos aquellos esfuerzos que busquen incrementar la comprensión de esta enfermedad en nuestro medio. Con tal fin, se describen tres casos de síndrome de linfohistiocitosis hemofagocítica secundaria. Esta es una complicación extremadamente infrecuente y poco sospechada; dos de los casos presentan complicación de linfomas no Hodgkin variedades T anaplásico y difuso de células grandes B rico en linfocitos T, y el tercero, complicación de una endocarditis infecciosa.
Casos clínicos
Caso 1
Paciente masculino de 41 años de edad y color de la piel negra que ingresó para estudio de síndrome febril de tres meses de evolución. Presentaba antecedentes de hepatitis B que adquirió por trasmisión vertical, enfermedad que estaba controlada al ingreso. Al examen físico destacaba hepatoesplenomegalia sin adenopatías periféricas. Los datos de laboratorio se muestran en la tabla 1. En la ecografía abdominal se observaron múltiples adenopatías en hilio hepático, peripancreáticas y periaórticas.
Tabla 1 Estudios complementarios realizados durante estadía hospitalaria
| Complementarios | Rango de referencia para adultos* | Pacientes | ||
|---|---|---|---|---|
| 1 | 2 | 3 | ||
| Hemoglobina(g/dL) | 12-16 | 8 | 9 | 6 |
| Leucocitos (por mm3) | 4500-1100 | 2500 | 1400 | 1000 |
| Plaquetas (por mm3) | 150-400 | 90 | 85 | 230 |
| Glucemia (mmol/L) | 4,21-6,11 | 4,5 | 2,5 | 3,7 |
| Creatinina (mmol/L) | 47,6 - 113,4 | 85 | 76 | 47 |
| Ácido úrico (mmol/L) | 156-357 | 270 | 300 | 230 |
| Proteínas totales (g/L) | 60-80 | 75 | 68 | 52 |
| Albúmina (g/L) | 35-52 | 38 | 24 | 18 |
| Colesterol total (mmol/L) | 3,87-6,71 | 3,2 | 1,5 | 2,8 |
| Triacilglicéridos (mmol/L) | 0,46-1,60 | 3,0 | 3,5 | 3 |
| Alanino amino transferasa (U/L) | 10-49 | 78 | 47 | 69 |
| Lactate deshidrogenasa (U/L) | 200-400 | 900 | 750 | 580 |
| Ferritina sérica (µg/L) | 30-400 | 600 | 750 | 590 |
| Tiempo de protrombina(s) | Control-12 Paciente-14 | Control-12 Paciente-17 | Control-12 Paciente-20 | |
| Tiempo de tromboplastina parcial activado(s) | Control- 30 Paciente-32 | Control-31 Paciente-47 | Control-32 Paciente-50 | |
| Fibrinógeno (g/L) | 2 - 4 | 1 | 1 | 0,7 |
* Intervalos de referencia usados en el Hospital Universitario Clínico Quirúrgico “Arnaldo Milián Castr”. Villa Clara, Cuba.
La presencia de fiebre de origen desconocido, pancitopenia y hepatoesplenomegalia con adenopatías intrabdominales conduce a la sospecha de enfermedad linfoproliferativa, por lo que se realiza medulograma en el que se evidenció fenómeno de hemofagocitosis (Fig. 1A). Con estos datos se plantea el diagnóstico de LHH secundaria (Tabla 2). Sin embargo, el paciente presentó coagulación intravascular diseminada irreversible a pesar de las medidas de soporte, falleciendo en el quinto día de estadía hospitalaria. El estudio de necropsia demostró como causa básica de muerte: linfoma no Hodgkin de células T anaplásico.
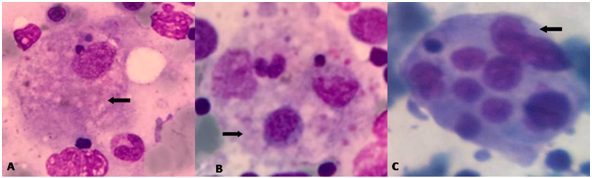
Tabla 2 Criterios para el diagnóstico de linfohistiocitosis hemofagocítica
| Criterios | Pacientes | ||
|---|---|---|---|
| 1 | 2 | 3 | |
| Historia familiar o diagnóstico genético específico | - | - | - |
| Fiebre mayor o igual a 38 oC durante 7 días o más | x | x | x |
| Esplenomegalia que rebase más de 3 cm el borde costal | x | x | x |
| Dos o más citopenias: hemoglobina (Hb) inferior a 9 g/dL; plaquetas por debajo de 100 000/mm3; polimorfonucleares neutrófilos por debajo de 1 000/mm3 | x | x | x |
| Hipertrigliceridemia (> 3 mmol/L) o hipofibrinemia (< 1,5 g/L) | x | x | x |
| Hemofagocitosis medular, líquido cefalorraquídeo o ganglios linfáticos sin evidencia de malignidad | x | x | x |
| Ferritina superior a 500 mg/L | x | x | x |
| CD25 soluble superior a 2 400 U/mL | - | - | - |
| Actividad de células asesinas naturales (NK) nula o disminuida | - | - | - |
| Número de criterios cumplidos | 6 | 6 | 6 |
El diagnóstico se realiza con cinco criterios de validación sobre ocho. El diagnóstico molecular (genes PRF, SAP, MUNC13-4, STX11, etcétera.) de una hemofagocitosis linfohistiocítica es suficiente para el diagnóstico.15
Caso 2
Paciente femenina, blanca, de 56 años de edad y antecedentes de salud, acudió a servicio de urgencias por cuadro febril prolongado de un mes de evolución, asociado a anorexia y pérdida de peso de alrededor de 8 kg en el último mes. Al examen físico se constató palidez cutáneo mucosa, hepatoesplenomegalia y adenopatías periféricas de aproximadamente 3 cm de diámetro, no dolorosas, móviles, de consistencia gomosa, localizadas en ambas cadenas cervicales laterales, axilares e inguinales. Estos datos en conjunto con anemia (Tabla 1) conducen a la realización de excéresis y biopsia de adenopatía cervical por la sospecha clínica de linfoma.
El estudio histológico reveló: linfoma no Hodgkin de células grandes B rico en linfocitos T CD20, CD3, CD5, CD79 positivos. Se realizó además medulograma y biopsia de médula ósea en los que no se evidenció signos de infiltración tumoral; pero se observaron abundantes macrófagos con fenómeno de hemofagocitosis (Fig. 1B). Al cumplir los criterios diagnósticos de LHH (Tabla 2) se inició tratamiento con Inmunoglobulina G endovenosa en dosis de 400 mg/kg de peso por cinco días y dexametasona en dosis de 10 mg/m2 superficie corporal diariamente con dosis descendente cada 2 semanas. A las dos semanas de iniciado este tratamiento la paciente presentaba mejoría clínica evidente y no existían signos de insuficiencias orgánicas por lo que se decidió administrar esquema de quimioterapia R-CHOP (rituximab, ciclofosfamida, vincristina, adriamicina, prednisona). A los 12 meses de seguimiento la paciente se encuentra en remisión completa de ambas condiciones.
Caso 3
Paciente femenina de 63 años de edad, con antecedentes de salud, acudió a cuerpo de guardia con astenia progresiva y fiebre de 39oC de dos semanas de evolución. El examen físico se constató palidez cutánea mucosa, tejido celular subcutáneo infiltrado hasta las rodillas de manera bilateral con edema blando, blanco, frío y de fácil Godet. La paciente presentaba crepitantes en ambas bases pulmonares y la frecuencia cardíaca era de 110 latidos por minuto.
Se auscultó soplo sistólico V/VI en foco tricuspídeo. El hallazgo de hepatoesplenomegalia al examen físico condujo a la realización de ecografía abdominal que confirmó la presencia de vísceromegalia y de adenopatías periaórticas y en el hilio esplénico.
Ante la sospecha clínica de endocarditis infecciosa se indicó ecocardiograma transtorácico que evidenció masa vegetante de 19 mm x 12 mm en valva tricuspídea nativa; sin embargo, los hemocultivos en medios aerobios no mostraron crecimiento bacteriano.
Los resultados de laboratorio de la paciente se muestran en la tabla 1. En la lámina periférica se destaca la presencia de punteado basófilo, policromatofilia y anillos de Cabot, en correspondencia con patrón hemolítico e índice reticulocitario de 7 %. La prueba de Coombs directa resultó positiva. El estudio del aspirado de médula ósea mostró hiperplasia de los sistemas megacariopoyético, granulopoyético y eritropoyético, se observaron numerosos macrófagos con fenómeno de hemofagocitosis (Fig. 1C).
Estos datos permitieron establecer los diagnósticos de:
Endocarditis infecciosa aguda de válvula tricúspide nativa con hemocultivos negativos;
Anemia hemolítica autoinmune secundaria a endocarditis infecciosa;
LHH secundaria a endocarditis infecciosa (Tabla 2).
El manejo terapéutico de esta paciente se basó en el uso de inmunoglobulina G endovenosa en dosis de 150 mg/kg asociado a metilprednisolona 200 mg diarios por cinco días, estímulo de la eritropoyesis con eritopoyetina recombinante y se inició tratamiento antibiótico con ciprofloxacino 400 mg cada 12 h, gentamicina 3 mg/kg/día y vancomicina 15 mg/kg cada 12h. Con este tratamiento la paciente logró estabilidad clínica en 21 días, lo que posibilitó el tratamiento quirúrgico de la lesión vegetante. Hasta el momento de la redacción de este artículo la paciente se encuentra en remisión clínica de ambas entidades.
Discusión
El escenario patogénico extremadamente amplio del síndrome de LHH, en donde los defectos genéticos, las enfermedades predisponentes y los factores desencadenantes de diferentes etiologías se encuentran mezclados, en conjunto con la alta tasa de mortalidad, hacen de esta enfermedad una de las situaciones clínicas más complejas y potencialmente mortales que existe.1,5,13
Por tal motivo, el diagnóstico y tratamiento precoz son la clave para mejorar los resultados en estos pacientes. Sin embargo, el diagnóstico de esta entidad y de los procesos patológicos que le subyacen continúa siendo un reto para los clínicos.2,4,14
Muchas de las manifestaciones clínicas de este síndrome son poco específicas. La combinación de síntomas, signos y hallazgos de laboratorio son similares a múltiples enfermedades que pueden ser desencadenantes u ocurrir simultáneamente al síndrome de LHH. El reto mayor consiste en identificar el momento de transición al síndrome de LHH, para lo cual son necesarias futuras investigaciones que ayuden a identificar el valor diagnóstico y pronóstico de distintos biomarcadores.4
El diagnóstico del síndrome de LHH se establece por la presencia de cinco de los ocho criterios diagnósticos aceptados actualmente y propuestos por la Sociedad Histiocítica-2004.15) Los tres pacientes presentados cumplieron 6 de los 8 criterios establecidos, siendo común a todos la identificación del fenómeno de hemofagocitosis en médula ósea (Fig. 1).
Los pacientes con cáncer son propensos a presentar el síndrome de LHH, principalmente aquellos con neoplasias hematológicas, especialmente linfomas.1,2,3 En este grupo de pacientes, cuando no se encuentran desencadenantes infecciosos, la causa del desarrollo del síndrome de LHH se ha relacionado con una excesiva secreción de citocinas por las células tumorales.1
El diagnóstico del síndrome de LHH asociado a linfoma puede ser especialmente difícil, ya que los síntomas constitucionales debidos al linfoma pueden ser atribuidos a este síndrome. En estos casos, el deterioro clínico puede ser muy rápido y los pacientes frecuentemente mueren poco tiempo después del diagnóstico, antes de que le sean administrados tratamiento efectivo para la cáncer subyacente.2 Por tal motivo, en pacientes adultos en los que se presente el síndrome de LHH y no se conozca la causa, se debe realizar una investigación exhaustiva para excluir malignidad, siendo el linfoma la malignidad más prevalente.2
En el caso 1 se presentó un paciente con factores claves de mal pronóstico: sexo masculino, enfermedad subyacente neoplásica y fallo en el diagnóstico y tratamiento de la enfermedad de base; el cual tuvo una evolución desfavorable de manera rápida.5 Otro aspecto a destacar de este caso es la poco frecuente presentación de hepatitis B, linfoma no Hodgkinde células T anaplásico y síndrome de LHH en un mismo paciente. En la literatura consultada no se encontraron informes de esta asociación. En este sentido, el síndrome de LHH ha sido identificado en pacientes con infecciones crónicas por virus de la hepatitis B y C, por lo que el mismo virus puede actuar tanto como factor predisponente como desencadenante de la activación macrofágica.1
La paciente presentada en el caso 2 tuvo un diagnóstico precoz de linfoma no Hodgkin de células grandes B rico en linfocitos T y en consecuencia recibió tratamiento efectivo para ambas enfermedades, con evolución satisfactoria. Estos dos casos se resalta la necesidad de un manejo intensivo en adultos en los que se sospecha el síndrome de LHH asociado a linfoma.
Existe un limitado número de casos reportados de síndrome de LHH vinculados a endocarditis infecciosa.16,17 En ellos los agentes infecciosos identificados han sido Aspergillus,18) Histoplasmacapsulatum,19) Mycobacterium tuberculosis,20 ) Estreptococos del grupo G,16) Haemophilusparainfluenzae,17) Abiotrophiadefective y Staphylococcus epidermidis.19) La paciente presentada en el caso 3 constituye el primer caso reportado en Cuba con la infrecuente asociación de estas dos entidades.
Este artículo, a pesar de solo reportar tres pacientes, constituye la serie más grande de pacientes cubanos publicada hasta la fecha. Los casos presentados destacan la gran complejidad del síndrome de LHH en el adulto y refuerzan la necesidad crítica del diagnóstico y tratamiento oportuno en estos pacientes. Debido a la baja incidencia de esta enfermedad y lo imperioso de tomar decisiones diagnósticas y terapéuticas en estos pacientes basadas en la evidencia nacional, se considera imprescindible promover esfuerzos multidisciplinarios y multicéntricos que logren la creación de un registro nacional de pacientes con síndrome de LHH.

