










 Serviços customizados
Serviços customizados Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroducción
Los síndromes mielodisplásicos (SMD) presentan un grupo heterogéneo de células progenitoras hematopoyéticas anormales en la médula ósea que conllevan un riesgo variable de evolución a neoplasias mieloides.1,2
La presencia concomitante de mieloma múltiple (MM) y SMD es rara e implica un desorden clonal en la maduración de las células plasmáticas.3
El MM es causado por un número excesivo de anomalías genéticas en las células plasmáticas o sus precursores (plasmoblastos). Tales anomalías se deben a una gran cantidad de eventos genéticos incluidas, mutaciones, polimorfismos de un sólo nucleótido, deleciones y duplicaciones de las regiones de un gen, una región o incluso un brazo completo de un cromosoma; translocaciones cromosómicas y variaciones en la expresión de genes intactos, entre otros.4
La inmunoglobulina M (IgM) es una proteína de membrana presente en la maduración de los linfocitos B y suele representar un fenotipo agresivo cuando se expresa en MM, se asocia generalmente con pronóstico desfavorable y una esperanza de sobrevida más corta que la de los pacientes con mieloma típico.5 En el presente trabajo se reportó un caso de MM en paciente con antecedentes de SMD con el objetivo de determinar las características clínicas y sus asociaciones con la expresión inmunofenotípica de inmunoglobulina M de superficie e inmunohistoquímica de CD20.
Presentación de caso
Paciente de sexo femenino de 67 años de edad. Admitida por pancitopenia en el servicio de hematología de un Hospital de Nivel III de Chiclayo-Perú. Con antecedentes de discrasia de células plasmáticas que representaron 18 % de la celularidad evaluada en el frotis de médula ósea. Al momento de la consulta la paciente presentó palidez ++/+++; ausencia de equimosis, petequias o sangrado activo.
Exámenes auxiliares
Hemoglobina (Hb): 8.9 g/dL, Volumen corpuscular medio: 108 fL, Leucocitos: 5.15 x 103/µL, Plaquetas: 105 x 103/µL, Ácido úrico: 7.5 mg/dL, Creatinina: 0.67 mg/dL, Urea: 40 mg/dL, Glucosa: 100 mg/dL, Lactato deshidrogenasa: 401 mg/dL, IgA: 12 md/dL, IgM: 9 md/dL, IgG: 519 md/dL, Bilirrubina total: 0.75 mg/dL, Calcio sérico: 10.1 mg/dL, β2-microglobulina: 7.21 µg/mL, Prueba de Coombs: negativo, TGO: 49 U/L, TGP: 96 U/L, TSH: 3.31 µUI/ml, T4 libre: 1.36 ng/dL, Examen de orina completo: normal.
Citometría de flujo de la médula ósea
El día 22/07/2019 se apreció un patrón madurativo aberrante en los granulocitos neutrófilos, monocitos y serie eritroide; con escaso número de precursores CD34+:0.15 %, sugerente de SMD (Fig. 1).
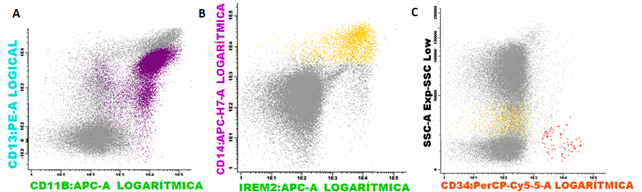
Fig. 1 Poblaciones aberrantes de neutrófilos y monocitos en síndrome mielodisplásico. Se aprecia un patrón aberrante de A. neutrófilos en color purpura; B. monocitos en color amarillo. Escaso número de C. precursores CD34+ en rojo.
El día 06/09/2019 se detectó una población, que representó 6 % de la celularidad total, CD38++, CD56+, CD19 negativo, CD45 negativo, CD117+, sIgM+: compatible con células plasmáticas anómalas (Fig. 2).
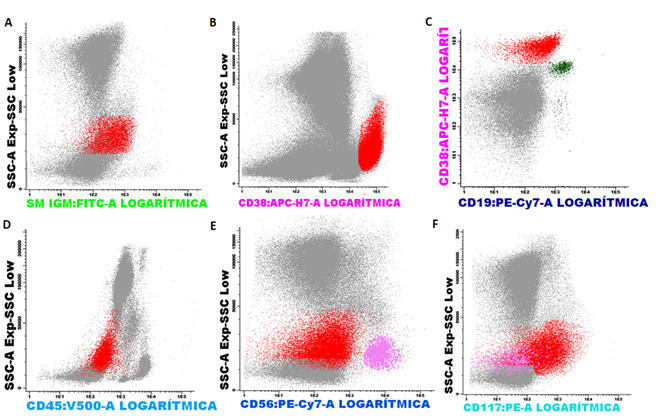
Discusión
La frecuencia de mieloma múltiple con la aberración inmunofenotípica sIgM+ es rara. Al igual que en los pacientes con MM sin este marcador, puede exhibir niveles altos de células plasmáticas monoclonales en la médula ósea más una inmunoglobulina circulante secretada por células plasmáticas malignas, ya sea IgG, IgA, IgM y expresión de una cadena ligera o ninguna.6,7
Los pacientes con MM también son propensos a presentar crisis de hipercalcemia, es decir, un episodio potencialmente mortal de niveles altos de calcio iónico (Ca2 +) en la sangre, debido a un exceso de reabsorción ósea y/o falla renal, así mismos niveles elevados de lactato deshidrogenasa sérica y Beta-2 microglobulina; en el presente caso destacó un incremento de este último.5
Los componentes monoclonales (M) parecen ser productos anormales de un único clon que presenta una proliferación excesiva. Algunos componentes M muestran actividad de anticuerpo, dirigida con mayor frecuencia contra autoantígenos y antígenos bacterianos. Los análisis más recientes sugieren que la expresión de los genes de las inmunoglobulinas que conducen a la producción de los componentes M tiene lugar de manera antigénicamente dirigida.3 Así mismo el deterioro de la producción de inmunoglobulina M en el mieloma múltiple puede deberse a la presencia de monocitos o macrófagos que dirigen la maduración de las células B normales hacia células plasmáticas secretoras de otros anticuerpos.8
En las células clonales con alta producción de IgM se desconoce la célula específica de origen. Sin embargo, el análisis de las secuencias de los genes de las inmunoglobulinas y de los marcadores de superficie celular sugiere la existencia de una transformación maligna de una célula del centro posgerminal.
Es importante señalar la presencia de CD20 en el presente caso, característico de la maduración de linfocitos B pequeños.9 Es probable que la paciente haya evolucionado hacia un mieloma y no hacia una leucemia mieloide, como suele suceder en las mielodisplasias, porque no existió exceso de blastos.10) Existe evidencia que la displasia en monocitos concomitante a MM puede asociarse a menor sobrevida libre de progresión.11) Lo que no puede corroborarse con el presente caso por el corto periodo de seguimiento, ya que la paciente se recuperó con terapia convencional y trasplante autólogo de células madre hematopoyéticas y en la actualidad se encuentra con buena salud.
Por otra parte, existe evidencia que las células estromales (CEM) de pacientes con SMD y MM presentan alteraciones funcionales similares a través de mecanismos moleculares comunes para inducir la senescencia celular, la muerte celular y la neovascularización. Específicamente las CEM presentan una proliferación deficiente in vitro, asociada a la sobreexpresión de CDKN2A y una regulación a la baja de CXCL12.
La inmunohistoquímica de la biopsia de la médula ósea ha demostrado que las CEM que no lograron proliferar in vitro y que expresan CDKN2A se acumulan intensamente en las regiones perivasculares, curiosamente, las CEM de los pacientes revelan una actividad de proliferación mejorada después del bloqueo de CDKN2A. 12Toda esta evidencia podría explicar la presencia concomitante de ambas enfermedades en el presente caso, ya que al no existir historia de tratamientos previos para ninguna de las dos, no podría haberse originado por la terapia de una de ellas y más alejado aún por un precursor común , debido a que como sabemos la célula plasmática y las células de linaje mieloide derivan de diferentes precursores.
La importancia del presente caso radicó en el hecho de detectar a tiempo y monitorear casos complejos de asociaciones inusuales de síndrome mielodisplásico y mieloma múltiple, con énfasis especial en el uso de la citometría de flujo para la evaluación del inmunofenotipo en neoplasias hematológicas, muestra un valor por encima de las técnicas morfológicas convencionales, lo que permite que el paciente sea oportunamente tratado.

