










 Serviços customizados
Serviços customizados Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroducción
La Enfermedad de Stargardt, o fundus flavimaculatus, es la distrofia macular juvenil más frecuente, responsable de 7 % de las distrofias maculares.1,2 Fue descrita en 1909, por el alemán Karl Bruno Stargardt,1) cuyos afectados presentaban atrofia macular, reducción progresiva y grave de la visión central, asociada a pequeñas manchas amarillentas profundas, típicamente en la primera y segunda décadas de la vida.2,3 En 1963, el termino fundus flavimaculatus fue introducido por Franceschetti,4 en el que describe un cuadro similar en el que las manchas amarillentas se extienden hacia la retina periférica, la cual aparece sobre todo en el adulto joven.5,6,7,8
Teniendo en cuenta la baja frecuencia de la enfermedad y que se trata de una distrofia retiniana que se precisa de más estudio para mayor comprensión, nos propusimos el objetivo de describir el proceso diagnóstico de una entidad poco común con forma de presentación infrecuente.
Presentación del caso
En la consulta de oftalmología del Hospital Docente Clínico Quirúrgico “Comandante Manuel Fajardo” se atendió una paciente de 21 años de edad, color de la piel blanca, quien asiste a consulta refiriendo “desde hace 5 años atrás he venido perdiendo la visión, pero he notado que en los últimos 4 meses mi situación visual ha empeorado casi no puedo leer y veo muy borroso”.
Al examen físico oftalmológico de su consulta inicial, presentaba una agudeza visual corregida de 20/80 en ojo derecho (OD) y de 20/50 en ojo izquierdo (OI) y una capacidad visual que no mejora con agujero estenopeico, presión intraocular de 15 mm hg en ambos ojos (AO). En la biomicroscopía del segmento anterior no se detectaron alteraciones en AO. Se realiza oftalmoscopía binocular indirecta en la que se evidenciaron lesiones retinianas hiperpigmentadas ovaladas con aspecto moteado, rodeada por una imagen blanquecina con aspecto “en baba de caracol”, por fuera de las arcadas temporales se observan lesiones pequeñas, puntiformes hipopigmentadas, diseminadas. En el examen físico de la consulta actual encontramos:
Examen físico ocular:
Ambos ojos:
Anexos: Sin alteraciones.
Segmento anterior: Sin alteraciones.
Medios: Transparentes.
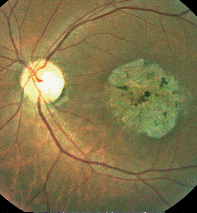
Fondo de ojo: (Oftalmoscopía directa e indirecta). De color rojizo en el que se observan las mismas lesiones ya descritas en la consulta inicial. (Figura 1).
Agudeza visual sin corrección:
OD: Cuenta dedos
OI: Movimientos de mano
Agudeza visual con corrección (VAP):
OD: -2.00 esf - 0.75 x 85º AV: 0.15
OI: -1.50 esf - 0.50 x 90º AV: 0.15
Test de visión al color (Ishihara): Patológico.
OD: 1/21
OI: 4/21
En la angiografía fluoresceínica se observó papila de aspecto normal, silencio coroidal en las primeras fases del examen y algunas lesiones pisciformes hiperfluorescentes. (Figura 2).
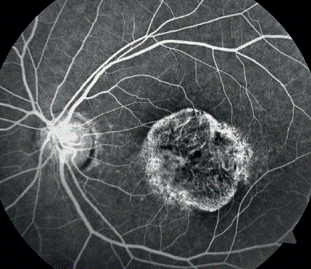
Fig. 2 Angiografía fluoresceínica papila normal, silencio coroidal en las primeras fases del examen y algunas lesiones pisciformes hiperfluorescentes.
Se le realiza una tomografía de coherencia óptica (OCT) que mostró disminución del espesor de la retina en la región macular en AO. En el electroretinograma, se registró disminución de la amplitud de la respuesta en todas sus fases.
La paciente evolucionó en un intervalo de 4 meses con empeoramiento de la agudeza visual, alcanzó una visión cuenta dedos en OD y movimiento de mano en OI, se realiza OCT evolutivo y se observa mayor disminución del espesor en la región macular en AO. Se interconsulta el caso con el servicio de baja visión para tratamiento con ayudas ópticas.
DISCUSIÓN
Las distrofias maculares pueden ser difíciles de abordar por diversos motivos. El diagnóstico diferencial a veces es difícil, y varias de las enfermedades producen ceguera legal a una edad relativamente joven.
El fundus flavimaculatus y la Enfermedad de Stargardt son variantes de una misma enfermedad heredadas de forma autosómica recesiva, ocasionadas por una mutación en un mismo gen ABCA4,7,9,10,11 responsable este del correcto funcionamiento del mecanismo de fototransducción, así como de la eliminación de los subproductos tóxicos derivados del metabolismo del ciclo de la visión, el mal funcionamiento de este gen conduce al depósito de lipofuscina y la atrofia del epitelio pigmentario de la retina, seguido de la degeneración de los fotorreceptores sobre todo en la zona macular, lo que provoca una pérdida de la visión central.8,9,12,13
La edad de inicio y las características clínicas iníciales de la Enfermedad de Stargardt son bastante variables; a veces, incluso, en personas de la misma familia. Un paciente puede tener pérdida de visión y cualquier combinación de la tríada clínica de atrofia macular, manchas y coroides oscura. Los signos que no estén presentes en la consulta inicial pueden aparecer posteriormente en la evolución del trastorno. Aunque en la mayoría de los pacientes el trastorno es lentamente progresivo, la expresividad puede variar desde una distrofia de los conos-bastones leve a una distrofia progresiva en la que hay un escotoma central creciente con el paso del tiempo.
Para establecer el diagnóstico en este paciente de Enfermedad de Stargardt con fundus flavimaculatus se tuvo en cuenta la edad de presentación de la enfermedad que fue alrededor de los 16 años de edad con afectación de la visión central bilateral, así como los hallazgos al examen físico (oftalmoscopía indirecta) se encontraron lesiones retinianas hiperpigmentadas ovaladas con aspecto moteado, rodeada por una imagen blanquecina con aspecto “en baba de caracol”; llama la atención la presencia por fuera de las arcadas temporales, lesiones redondas, pequeñas, puntiformes amarillentas, hipopigmentadas, diseminadas por el polo posterior y coloración rojiza del fondo de ojo que nos hace pensar en un fundus flavimaculatus; este último se presenta en la edad adulta, no siendo frecuente que se encuentren estas características en un mismo paciente.14,15,16
Debemos resaltar la importancia de las pruebas diagnósticas como la angiografía fluoresceínica en la que se observó el silencio coroideo en las primeras fases, así como algunas lesiones pisciformes hiperfluorescentes, el OCT y el electrorretinograma descritos ya anteriormente. No se presentó ningún tipo de dificultad al realizar las pruebas diagnósticas, pues constamos con el equipamiento necesario en nuestro medio. El diagnóstico clínico de Enfermedad de Stargardt se confirma por el hallazgo de una “coroides oscura” en la angiografía con fluoresceína. Este fenómeno, en el que la circulación retiniana resalta sobre una coroides hipofluorescente, está presente en, al menos, 80 % de los pacientes con el trastorno. Aunque la ausencia de este signo no descarta la Enfermedad de Stargardt, su presencia es bastante específica de esta. Se cree que el signo de la coroides oscura representa el enmascaramiento de la fluorescencia coroidea por una acumulación de pigmento similar a lipofuscina en todo el epitelio pigmentario de la retina.9,13,17,18 Las numerosas lesiones hiperfluorescentes que se evidencia en la angiografía en pacientes con manchas parecen representar defectos de la transmisión alrededor de las manchas.
El diagnóstico diferencial de la Enfermedad de Stargardt incluye las entidades que pueden producir una maculopatía atrófica, entre ellas la Enfermedad de Best o distrofia viteliforme macular de herencia autosómica dominante en la que la agudeza visual puede permanecer inalterada hasta fases muy avanzadas; se pueden observar mediante la oftalmoscopía indirecta lesiones subrretinianas en forma de yema de huevo, seudohipopión en caso de reabsorción parcial, neovascularización coroidea y cicatrización disciforme con un electroretinograma normal y un electrooculograma alterado, fundus albipuntactus, retinosis puntata albescens y drusen de la membrana de Bruch.2,6,7 Por otra parte, tenemos la distrofia macular y la miopía magna, esta de igual manera con un patrón de herencia autosómica dominante, en la que existe miodesopsias y escotoma central, al examen fundoscópico podemos encontrar grandes capas de atrofia en el polo posterior, atrofia circumpapilar, estafiloma posterior, manchas de fuchs, además de estrías lacadas y neovascularización coroidea.15,16,17,19
En la actualidad, los procesos fisiopatológicos que producen pérdida visual en estas enfermedades no pueden detenerse ni revertirse; sin embargo, es posible asegurar a los pacientes que la enfermedad progresa lentamente y que siempre mantendrán alguna visión útil.17,18,19) Aunque hoy, no existe ningún tratamiento efectivo para los pacientes con Enfermedad de Stargardt, se puede decir que se están desarrollando diferentes aproximaciones terapéuticas muchas de ellas ya en proceso de ensayo clínico lo cual nos sitúa más cerca de encontrar un tratamiento que sea efectivo y evite la pérdida de visión en los centenares de niños y jóvenes que se diagnostican cada año con esta enfermedad.
Teniendo en cuenta que se trata de una forma atípica de esta afección donde se presentaron las dos variantes de una misma enfermedad, las cuales son causa de disminución de la visión de forma bilateral y de progresión rápida, y después de obtener los datos descritos en el examen físico ocular de la paciente se puede plantear que el pronóstico visual del paciente es reservado.
Conclusiones
La mayoría de las distrofias retinianas tiene, desde el punto de vista clínico, sus semejanzas, en cambio su evolución y pronóstico pueden ser diferentes. Por lo que es de gran importancia conocer sus características clínicas, epidemiológicas, así como los exámenes o pruebas diagnósticas que contribuyen a la confirmación del diagnóstico, así como un adecuado consejo genético y pronóstico de su condición.

