










 Serviços customizados
Serviços customizados Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La proteína quinasa CK2 se encuentra frecuentemente desregulada en células tumorales y es responsable del 20 % del fosfoproteoma celular. 1 Esta quinasa se vincula con rasgos característicos del cáncer como la proliferación exacerbada, supervivencia celular, inhibición de la apoptosis, angiogénesis, y metástasis. 2 Se han descrito hasta el momento más de 300 sustratos celulares para CK2 y modula la función de los supresores tumorales PTEN y PML, así como los oncogenes AKT y c-myc. 3-6 De igual forma, la expresión aberrante de CK2 impacta en múltiples cascadas de señalización cuya desregulación conlleva a la transformación maligna las cuales incluyen Wnt, Hh, JAK/STAT, y PI3K/AKT. 7
La actividad enzimática de CK2 se lleva a cabo por las subunidades catalíticas CK2( y CK2(’ por separado o unidas con el dímero de subunidades reguladoras CK2( para formar la holoenzima. 8 La fosforilación de los llamados sustratos clase III puede ser solo ejercida por la holoenzima, mientras los sustratos clase I pueden ser fosforilados por la holoenzima o las subunidades catalíticas individuales. 9 Los sustratos clase II son solo fosforilados por las subunidades catalíticas de CK2. 9
La CK2 es actualmente considerada como un blanco promisorio y “drogable” en cáncer debido a las exitosas pruebas de concepto in vitro e in vivo usando moléculas químicas pequeñas inhibidoras de la actividad enzimática mediante el bloqueo del sitio de unión al ATP en la subunidad catalítica CK2( o por inhibición de la interacción entre las subunidades para formar la holoenzima. 10,11 De igual forma el uso de oligonucleótidos antisentido contra la CK2( ha servido para validar la perspectiva de esta enzima como blanco para el tratamiento del cáncer. 12
El CIGB-300 es un péptido que actualmente se evalúa en la clínica y forma parte de la cartera de nuevos desarrollos farmacéuticos anticáncer del CIGB. Es un inhibidor de la fosforilación mediada por CK2 a través de su unión directa y bloqueo del dominio fosfoaceptor conservado en los sustratos para dicha enzima. 13 Evidencias experimentales en líneas celulares de tumores sólidos indicaron que la proteína B23/NPM1 era un sustrato de CK2 mayoritariamente unido por el CIGB-300 en ensayos de pull-down in vivo. 14 Igualmente en lisados celulares el CIGB-300 une diferentes sustratos de CK2 y en células de leucemia linfocítica crónica se encontró que el CIGB-300 inhibió la fosforilación de otros sustratos como AKT y PTEN. 15,16
Teniendo en cuenta el mecanismo descrito para el CIGB-300 y que ha sido reportado un dominio fosfoaceptor en la subunidad CK2(, 17,18 nos propusimos investigar si el CIGB-300 es capaz de interaccionar directamente con la enzima e inhibir su actividad. Los resultados permitieron validar dicha hipótesis científica y además conocer el fosfoproteoma regulado por el CIGB-300 en células tumorales.
Como parte de la presente investigación nos propusimos además explorar la posible sinergia antineoplásica del CIGB-300 con quimioterapéuticos e inhibidores del receptor para el factor de crecimiento epidérmico (EGFR) brindando así una contribución a las bases moleculares que pueden sustentar la sinergia del CIGB-300 con esas drogas anticáncer.
De conjunto, el presente trabajo persigue como objetivo fundamental exponer nuevos elementos vinculados al mecanismo de acción antineoplásico del CIGB-300, relacionados con el modo de inhibición de dicha fosforilación y la sinergia del péptido con drogas anticáncer. De manera importante, los resultados obtenidos no solo tienen un impacto científico-técnico, sino que sustentan el uso racional del péptido CIGB-300 en esquemas terapéuticos combinados con la terapia estándar para el tratamiento del cáncer.
MÉTODOS
Ensayos radiométricos y de interacción
Las reacciones de fosforilación se realizaron con la holoenzima o la subunidad CK2( usando [γ-32P]-ATP (6000 Ci/mmol) en presencia del CIGB-300 y los péptidos sustratos M y 29, o los sustratos recombinantes GST-Olig2 y GST-Six1. La actividad enzimática se expresa como valores directos de conteos por minuto (CPM). La interacción entre las subunidades de CK2 y el CIGB-300 se realizó por incubación de GST-CK2( en ausencia o presencia del CIGB-300 (50 µM) y se adsorbió a una matriz de glutatión-sefarosa. Seguidamente se adicionó CK2( y se determinó la actividad enzimática. Para la interacción en solución, la CK2α se incubó por 30 min en presencia o ausencia del CIGB-300 (100 µM) y cantidades crecientes de CIGB-300 biotinilado (CIGB-300-B) adsorbido en matriz de estreptavidina-sefarosa. Finalmente, se realizó la reacción enzimática usando el péptido 29.
Ensayos de pull-down y colocalización in situ
Los ensayos de pull-down se realizaron usando el CIGB-300-B para capturar las diferentes proteínas de interés como CK2(, AKT, PTEN, o B23/NPM1 en los lisados celulares. Las proteínas adheridas al CIGB-300 se analizaron por Western blot. Para la microscopía fluorescente, las células NCI-H125 se trataron a diferentes tiempos con el CIGB-300-B. Después de la fijación y permeabilizaron las células se incubaron con anticuerpos anti-CK2(, anti-CK2( o anti-B23/NPM1. Finalmente, se añadió el conjugado avidina-FITC o el correspondiente anticuerpo secundario conjugado a Alexa Fluor 594 y el análisis se realizó en un microscopio Confocal Leica Microsystem.
Estudios de proteómica
El fosfoproteoma de CK2 regulado por el CIGB-300 se exploró en células NCI-H125 tratadas o no con el CIGB-300 durante 10 y 30 minutos. Los fosfopéptidos de los extractos celulares fueron aislados por enriquecimiento mediante adherencia a matriz de TiO2 y posteriormente sometidos a LC-MS/MS para su análisis de identidad. Para la proteómica comparativa, la combinación CIGB-300 (50 µM/ml) + Cisplatino (0,5 µM/ml) o los agentes por separados fueron incubados con las células NCI-H125 y los extractos nucleares preparados al final del tratamiento. Después del aislamiento proteico, la digestión tríptica y el marcaje isotópico diferencial se realizó la identificación y cuantificación de las proteínas mediante LC-MS/MS. Los resultados muestran el factor de cambio de los niveles de cada proteína.
RESULTADOS Y DISCUSIÓN
La investigación realizada en este trabajo se centra en responder 2 preguntas científicas esenciales dentro del mecanismo de acción del CIGB-300. En primer lugar, conocer si este péptido además de interactuar e inhibir el sitio fosfoaceptor de los sustratos de CK2 era capaz de inhibir también a la propia enzima, y en segundo lugar explorar el sinergismo del CIGB-300 con drogas anticancerígenas con un acercamiento a las bases moleculares que puedan sustentar dicha sinergia.
Para despejar un posible efecto directo del CIGB-300 sobre la enzima CK2 se realizaron ensayos radiométricos de fosforilación mediada tanto por CK2( (subunidad catalítica) como por la holoenzima (CK2( + CK2(), en presencia de dosis seriadas del CIGB-300 (figura 1). 19
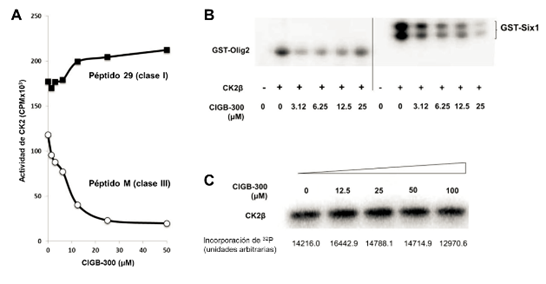
Fig. 1 Efecto del CIGB-300 sobre la actividad de la holoenzima CK2. Los ensayos radiométricos para la fosforilación por la holoenzima CK2 o la subunidad CK2( se realizaron usando 28 ng de estas y cantidades crecientes del CIGB-300. Las reacciones se llevaron a cabo en presencia 10 mM MgCl2 y 100 mM [γ-32P]-ATP (6000 Ci/mmol), así como 1 mM de los péptidos sustratos M, 29 (A), los sustratos recombinantes GST-Olig2, GST-Six1 (B), o la subunidad CK2( (C).
De manera interesante, al usar como sustrato al péptido M, el cual es fosforilado solo por la holoenzima CK2, se observó una marcada inhibición dosis dependiente de la actividad enzimática en presencia del CIGB-300 (figura 1a). De igual forma, otros 2 sustratos específicos para la holoenzima CK2 (GST-Olig2 y GST-Six1) fueron inhibidos también en presencia del CIGB-300 (figura 1b). Por el contrario, el péptido 29 y la calmodulina, los cuales son fosforilados fundamentalmente por las subunidades CK2(, no fueron inhibidos en presencia del CIGB-300. Tampoco la autofosforilación de la subunidad regulatoria de CK2( fue modificada en presencia de dosis seriadas del péptido lo cual sugiere que el CIGB-300 no está bloqueando ese sitio fosfoaceptor en particular (figura 1c).
De esta forma el CIGB-300 inhibe la actividad enzimática de la holoenzima CK2 sin interaccionar con la subunidad CK2(. De igual forma se realizaron ensayos radiométricos donde la CK2( se incubó con cantidades crecientes de CK2( en presencia o no del CIGB-300. Como esperado, la actividad enzimática de la holoenzima se incrementó de manera proporcional a la adición de la subunidad CK2( en la reacción, sin embargo, en presencia del CIGB-300 la actividad fue inhibida. 19 Sin embargo, el péptido interaccionó directamente con la subunidad CK2( tanto en solución como cuando se fijó a un soporte sólido (figura 2 A, B). 19
En aras de corroborar esta interacción en el contexto celular se realizaron ensayos de pull-down usando el CIGB-300 biotinilado para capturar la CK2( en lisados celulares. Los resultados de la figura 2c demuestran que el CIGB-300 se unió directamente a la CK2( con más de un 90 % de captura total tanto en lisados de células NCI-H125 como de HPB-ALL de leucemia linfocítica aguda. 19,20 La unión del CIGB-300 a la proteína B23/NPM1 fue usada como control positivo de interacción en estos ensayos (figura 2C). 14
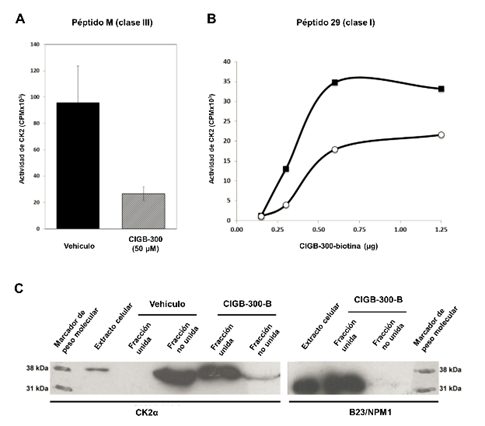
Fig. 2 Evaluación de la interacción del CIGB-300 con CK2(. La proteína de fusión GST-CK2( (80 ng) fue incubada en ausencia o presencia del CIGB-300 (50 µM). Una vez adsorbida a una matriz de glutatión-sefarosa se añadieron 160 ng de CK2( y se determinó la actividad enzimática usando el péptido M (A). Alternativamente, la CK2α (240 ng) fue incubada en presencia o ausencia de 100 µM del CIGB-300 y cantidades crecientes de CIGB-300-B adsorbido en matriz de estreptavidina-sefarosa. Después de varios lavados, se llevó a cabo la reacción enzimática con el péptido 29 (B). Para capturar la CK2( en los lisados celulares se realizaron ensayos de pull-down con CIGB-300-B adsorbido a una matriz de estreptavidina-sefarosa. Después de varios lavados, las proteínas adheridas al CIGB-300 se analizaron por Western blot usando anticuerpos específicos contra CK2( y B23/NPM1 (C).
Finalmente, para confirmar la proximidad física del CIGB-300 con la CK2( en el contexto celular se realizaron ensayos de colocalización in situ mediante microscopía de fluorescencia confocal en células NCI-H125. Los resultados demostraron una evidente colocalización del CIGB-300 con la subunidad CK2( en el citoplasma de las células con un aparente reforzamiento en el área perinuclear y más ligeramente dentro del área nuclear según indica la fluorescencia color naranja (figura 3A). 19 En cambio, la colocalización del CIGB-300 con B23/NPM1 usado como control positivo en esta experimentación, fue más evidente en el núcleo y nucléolos (figura 3B).

Fig. 3 Colocalización in situ del CIGB-300 con CK2α. Las células NCI-H125 se trataron con CIGB-300-B (50 μM) durante 10 min y seguidamente se lavaron, fijaron y permeabilizaron. Después del bloqueo con BSA (4 %) se incubaron con anticuerpo anti-CK2α (A) o anti-B23/NPM1 (B), este último usado como referencia de colocalización. Finalmente, los conjugados avidina-FITC, o anticuerpos secundarios conjugados a Alexa Fluor 594 fueron añadidos. El análisis se realizó en un microscopio de fluorescencia Leica Microsystem. La tinción con DAPI (azul) se realizó para contrastar el núcleo celular.
En línea con esta hipótesis hemos observado que el tratamiento de células HPB-ALL con CIGB-300 inhibió la fosforilación de 2 sustratos claves de CK2 como son AKT y PTEN sin mediar interacción directa del péptido con estos en ensayos de pull-down. 20 Este mecanismo dual de inhibición de la fosforilación mediante interacción tanto con el sustrato como con la holoenzima CK2 permiten robustecer la condición del CIGB-300 como compuesto “Primero de su Clase” y resulta un elemento distintivo respecto a otros inhibidores de CK2 y de otras quinasas. Por tal motivo, estos hallazgos constituyen parte esencial de la novedad científica de este trabajo.
Como parte de la investigación relacionada con la inhibición de la fosforilación mediada por CK2 del CIGB-300, en nuestro trabajo exploramos por primera vez el fosfoproteoma celular regulado tempranamente por este péptido. Los resultados permitieron conocer el perfil global de inhibición de la fosforilación por el CIGB-300, el cual se caracterizó por una gran disminución de la fosforilación de sitios que contenían Ser en un entorno de residuos ácidos, típico de sustratos de CK2. 19 La identidad de los sustratos de CK2 a los cuales pertenecían los fosfositios inhibidos por el CIGB-300 indicó que 8 y 10 sustratos para esta quinasa fueron inhibidos a los 10 min y 30 min, respectivamente. 19 Además, el análisis bioinformático indicó que los sustratos inhibidos están involucrados en procesos como la biogénesis ribosomal, el metabolismo, procesamiento de ARN, apoptosis, expresión génica y ciclo celular, lo cual pudiera contribuir al efecto antineoplásico in vitro e in vivo del CIGB-300.
Al integrar los resultados de este trabajo podemos concluir que el CIGB-300 inhibe un conjunto de sustratos de CK2 (tabla 1) y que en el contexto celular ambos mecanismos de inhibición de CK2 por el CIGB-300 pudieran operar de manera concomitante.
Tabla 1 Relación de sustratos de CK2 inhibidos por el CIGB-300
| Sustrato | Nombre | Sitio inhibido | Metodología empleada |
|---|---|---|---|
| B23/NPM1 | Nucleofosmina 1 | Ser125 |
|
| HDAC1 | Histona deacetilasa 1 | Ser393 | Fosfoproteómica 19 |
| PTEN | Fosfatidilinositol-3,4,5-trisfosfato 3 fosfatasa | Ser380 |
|
| AKT | Proteína quinasa B | Ser129 |
|
| EEF1D | Factor de elongación 1 delta | Ser162 | Fosfoproteómica 19 |
| USP7 | Ubiquitina carboxil-terminal hidrolasa A7 | Ser18 | Fosfoproteómica 19 |
| STMN1 | Estatmina 1 | Ser16/63 | Fosfoproteómica 19 |
| ABCF1 | Transportador ABC de la subfamilia F1 | Ser109 | Fosfoproteómica 19 |
| PDCD5 | Proteína de muerte celular programada 5 | Ser119 | Fosfoproteómica 19 |
| PPP1R2 | Inhibidor de proteína fosfatasa 2 |
Ser121/ Ser122 |
Fosfoproteómica 19 |
| SEPT2 | Septina 2 | Ser218 | Fosfoproteómica 19 |
| HMGA1 | Proteína del grupo de alta movilidad A1 |
Ser102/ Ser103 |
Fosfoproteómica 19 |
| CDC37 | Proteína co-chaperona de HSP90 | Ser13 | Fosfoproteómica 19 |
| MYH9 | Miosina 9 | Ser1943 | Fosfoproteómica 19 |
| HSP90AB1 | Proteína de choque térmico de 90 kDa | Ser255 | Fosfoproteómica 19 |
| HSP90AB2P | Proteína de choque térmico de 90 kDa beta 2 | Ser177 | Fosfoproteómica 19 |
La integración de los resultados nos permite proponer un modelo en el cual la inhibición de la holoenzima CK2 por el CIGB-300 se realiza por interacción directa con la subunidad CK2( dando lugar a una forma “no productiva” o inactiva de dicha holoenzima. Estos resultados también refuerzan la novedad científica del trabajo por ser la primera descripción del fosfoproteoma global regulado por el CIGB-300 en células tumorales y por otro lado por brindar un panorama más amplio de la inhibición que ejerce el CIGB-300 sobre la fosforilación mediada por CK2 en el contexto celular.
Como parte del mecanismo de acción antineoplásico del CIGB-300 se investigó además la capacidad del péptido de sinergizar con algunos quimioterapéuticos de amplio uso en oncología clínica e inhibidores del EGFR. En particular, exploramos la combinación in vitro del CIGB-300 con el cisplatino (agente alquilante), el paclitaxel (antimitótico), la doxorubicina (antitopoisomerasa II) y el 5-fluorouracilo (antimetabolito) en células humanas de cáncer de pulmón y de cuello uterino. 21
Para determinar el tipo de interacción (sinérgica, aditiva o antagónica) se realizó un diseño de cuadrado latino donde se mezclan diferentes dosis de cada droga y el efecto sobre la viabilidad celular es cuantificado y procesado por el programa Calcusyn el cual a partir de análisis dosis-respuesta de las combinaciones genera parámetros farmacológicos importantes como el índice de combinación (IC). Valores de IC menor e igual que de 0,5 indica fuerte sinergismo, de 0,5 a 0,9 sinergia/aditividad, igual a 1,0 aditividad y mayor que 1,0 antagonismo. De manera interesante se observó sinergismo del CIGB-300 con el paclitaxel y el cisplatino con valores de IC de 0,30 y 0,83, respectivamente. En cambio, para la doxorubicina y el 5-fluorouracilo, el análisis indicó muy escasa sinergia y áreas de ligero antagonismo para el rango de dosis investigado. Adicionalmente, se exploró in vivo la combinación del CIGB-300 con el Cisplatino en un modelo preclínico de cáncer. (21 Los resultados demostraron sinergismo de efecto antitumoral significativo entre ambas drogas en términos de supervivencia de los animales con tumor. Las combinaciones más relevantes fueron: CIGB-300 (50 µg) más cisplatino (1 mg/kg) y CIGB-300 (200 µg) más cisplatino (4 mg/kg).
Por su parte, los resultados de la combinación in vitro del CIGB-300 con inhibidores del EGFR indicaron sinergia del péptido con el erlotinib con valores de IC de 0,54 a 0,76 para células NCI-H460 y de 0,35 a 0,83 en células A549. (22 De manera similar se pudo apreciar un incremento del efecto citotóxico de la combinación del CIGB-300 con el nimotuzumab en las dosis evaluadas en células A431. (23) De conjunto podemos asegurar que el CIGB-300 no solo es capaz de incrementar su efecto antineoplásico combinado con quimioterapéuticos sino también en presencia de inhibidores del EGFR, aspecto que robustece la novedad científica de este trabajo.
Finalmente nos propusimos investigar las bases moleculares que sustentan el sinergismo entre el CIGB-300 y las drogas anticancerígenas. En este sentido, los resultados observados anteriormente que refieren inhibición de la fosforilación por CK2 en PTEN y AKT por el CIGB-300 sirven de soporte molecular para sustentar el sinergismo antineoplásico del CIGB-300 tanto con los quimioterapéuticos como con los inhibidores del EGFR, pues se conoce que dicho evento bioquímico en ambos sustratos juega un rol crucial en la resistencia tumoral. (24
Adicionalmente se exploró mediante proteómica comparativa el proteoma de células NCI-H125 en presencia de la combinación CIGB-300 más cisplatino y cada agente por separado. Como resultado se logró identificar un total de 28 proteínas vinculadas con la resistencia al cisplatino cuyos niveles resultaron modificados por la combinación de este con el CIGB-300 (tabla 2). (25
Tabla 2 Proteínas relacionadas con el fenotipo de resistencia al cisplatino que cambiaron sus niveles en presencia de la combinación con CIGB-300
| Uniprot_ID | Proteína | Expresión en células resistentes al cisplatino | |
|---|---|---|---|
| P00558 | Fosfoglicerato quinasa 1 | 0 | Elevada |
| P38919 | Factor eucarionte de iniciación 4A-III | 0 | Elevada |
| P22626 | Ribonucleoproteína heterogénea nuclear A2/B1 | 0,1 | Elevada |
| P09429 | Proteína de alta movilidad B1 | 0,1 | Elevada |
| Q09028 | Proteína de unión a histona RBBP4 | 0,1 | Elevada |
| P62888 | Proteína ribosomal 60S L30d | 0,1 | Elevada |
| P33991 | Componente del complejo de mantenimiento de minicromosomas 4 | 0,1 | Elevada |
| Q02952 | Proteína de anclaje de A-quinasa 12 | 0,1 | Elevada |
| Q15233 | Proteína NonO | 0,1 | Elevada |
| P08670 | Vimentina | 0,2 | Elevada |
| Q04695 | Citoqueratina 17 | 0,2 | Elevada |
| P08107 | Proteína de choque térmico de 70kDa 1A | 0,2 | Elevada |
| P21291 | Proteína rica en cisteína y glicina 1 | 0,2 | Elevada |
| P11142 | Proteína de choque térmico de 71 kDa | 0,2 | Elevada |
| P13073 |
Citocromo C oxidasa subunidad 4 (isoforma 1, mitocondrial) |
0,2 | Elevada |
| Q15149 | Plectina | 0,3 | Elevada |
| P60709 | Actina, citoplasmática 1 | 0,3 | Elevada |
| P0C0S5 | Histona H2A.Z | 0,3 | Elevada |
| P51858 | Factor de crecimiento derivado de hepatoma | 0,3 | Elevada |
| P07910 | Ribonucleoproteína heterogénea nuclear C1/C2 | 0,3 | Elevada |
| Q96PK6 | Proteína de unión al ARN 14 | 0,3 | Elevada |
| Q03252 | Lamina B2 | 0,3 | Elevada |
| P52272 | Ribonucleoproteína heterogénea nuclear M | 0,3 | Elevada |
| P09382 | Galectina 1 | 0,3 | Elevada |
| P04264 | Queratina 1 | 3,3 | Disminuida |
| P47756 | Proteína CapZ beta | 3,6 | Disminuida |
| P31946 | Proteína 14-3-3 beta/alfa | 8,2 | Elevada/Disminuida |
| Q15070 | Proteína de la membrana mitocondrial interna OXA1L | 42,3 | Disminuida |
FC: Factor de cambio
Como se ha descrito por otros autores el factor transcripcional NF-kB juega un papel esencial como mediador de la resistencia al Cisplatino y otros quimioterapéuticos. (26,27 Teniendo en cuenta tales antecedentes nos propusimos evaluar cómo impactaría el CIGB-300 en los niveles de dicho factor en el contexto de un fenotipo de resistencia adquirida al Cisplatino en células de cáncer de pulmón. Con ese propósito se generó, a partir de las células parentales A549, una sublínea denominada A549-cispR, las cuales desarrollaron un fenotipo resistente al Cisplatino mediante exposición crónica in vitro a esa droga. Los resultados obtenidos demostraron que la sublínea A549-cispR tenía niveles intrínsecos del factor NF-kB más elevados que la línea celular parental los cuales fueron reducidos por el CIGB-300. Además, la sensibilidad de esta línea al CIGB-300 fue mayor que la observada en la parental. (28
De conjunto, los resultados obtenidos aportan un grupo de eventos moleculares modulados por el CIGB-300 los cuales pudieran contribuir a las bases moleculares que soportan la sinergia y aditividad con otros quimioterapéuticos ampliamente usados en oncología clínica como el paclitaxel y el cisplatino, así como con inhibidores del EGFR como el erlotinib y el nimotuzumab, los cuales han probado demostrada eficacia en el tratamiento de varios tipos de tumores sólidos. Estos hallazgos pueden tener un impacto científico-técnico y social en cuanto a la posibilidad de nuevas opciones terapéuticas para el tratamiento del cáncer basadas en combinaciones del CIGB-300 con dichas drogas.
Conclusiones
Los aportes científicos generados de la presente investigación se pueden resumir de la siguiente forma:
El CIGB-300 interacciona directamente con la subunidad CK2( e inhibe la actividad enzimática de la holoenzima dando lugar a un mecanismo dual de inhibición no previamente descrito para otros inhibidores de CK2 y de otras proteínas quinasas.
El fosfoproteoma de CK2 regulado por el CIGB-300 se caracterizó por la inhibición de la fosforilación de varios sustratos de CK2 vinculados con procesos celulares relevantes en cáncer como la biogénesis ribosomal, el metabolismo exacerbado y procesamiento de ARN, la apoptosis, la expresión génica y ciclo celular.
El CIGB-300 sinergiza con el paclitaxel, el cisplatino, e inhibidores del EGFR, lo cual es un elemento de gran valor para la práctica médica en particular para el diseño de futuros estudios clínicos del CIGB-300 en cáncer.
La inhibición por el CIGB-300 de la fosforilación de PTEN y AKT, así como la reducción de los niveles de NF-kB y la modulación de un conjunto de proteínas vinculadas con la resistencia al cisplatino, constituyen las bases moleculares que pueden sustentar la sinergia del péptido con quimioterapéuticos y con inhibidores del EGFR.

