










 Serviços customizados
Serviços customizados Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroducción
Las enfermedades ampollosas autoinmunes representan un grupo heterogéneo de alteraciones poco comunes en un órgano específico, que resultan potencialmente fatales y afectan la piel y las membranas mucosas. Sus manifestaciones clínicas son diferentes, pero comparten parcialmente mecanismos inmunopatológicos, que están asociados con depósitos de autoanticuerpos y con el complemento contra diferentes moléculas de la epidermis y la unión dermoepidérmica.1
El pénfigo conforma un grupo de enfermedades dermatoampollosas intraepidérmicas de origen autoinmune, que se manifiestan por la separación entre las células epidérmicas. Este fenómeno es conocido como acantólisis y resulta en la formación de ampollas, erosiones en las membranas mucosas y costras en la piel.2
Dicha dermatopatía se clasifica en cuatro tipos: pénfigo vulgar, con su variante vegetante, en el que las ampollas se forman en la parte basal de la epidermis; pénfigo foliáceo y pénfigo eritematoso, caracterizados por ampollas que se encuentran en la epidermis apical; y pénfigo paraneoplásico, con ampollas en la parte suprabasal asociadas a neoplasias, como el linfoma maligno, el carcinoma broncogénico de células escamosas y la leucemia linfocítica crónica, entre otras.3,4
Al respecto, el pénfigo vulgar (PV) es la variante más común y grave, que comprende hasta 80 % de todos los casos de pénfigo. Aun así, se trata de una enfermedad rara, con una incidencia mundial de aproximadamente 0,5 casos por cada 100 000 habitantes. Posee una evolución crónica, con numerosos episodios de agudizaciones y remisiones, a pesar del tratamiento. Antes del uso de esteroides y de la terapia inmunosupresora, la mortalidad variaba entre 60 y 90 %, debido principalmente al choque séptico.4
Desde el punto de vista clínico, se distingue por la aparición de vesículas flácidas, que al romperse producen erosiones dolorosas, de forma y tamaño irregulares, con bordes poco definidos, cubiertas por costras hemorrágicas. En 50 a 70 % de los pacientes con pénfigo vulgar las lesiones aparecen en la mucosa bucal meses antes de extenderse al resto de la piel.5
Las manifestaciones oculares del PV son poco frecuentes y están generalmente limitadas a la conjuntiva, los párpados, o a ambos. La córnea rara vez suele estar afectada; en pocas publicaciones se describen casos de gravedad con úlceras corneales y perforación, a pesar del uso de la terapia inmunosupresora.6 En este artículo se presenta un caso poco común de úlcera corneal con perforación bilateral.
Caso clínico
Se trata de un anciano de 72 años de edad, fumador inveterado, que se encontraba ingresado en el Servicio de Dermatología del Hospital General Docente Dr. Juan Bruno Zayas Alfonso de Santiago de Cuba por el diagnóstico de pénfigo vulgar en sus inicios, para lo cual llevaba tratamiento con inmunosupresor y esteroides. Este acudió a la consulta de Oftalmología de la misma institución hospitalaria por presentar enrojecimiento ocular, unido a disminución de la visión y secreciones abundantes de aproximadamente 5 días de evolución.
Examen oftalmológico
En el ojo izquierdo y sus anexos se encontró hiperemia conjuntival bulbar, edema de párpados con costras amarrillas y secreción conjuntival amarrillo-verdosa. A su vez, existía retracción palpebral con vesículas palpebrales. En la biomicroscopia con lámpara de hendidura del segmento anterior se observó un infiltrado estromal en la región paracentral inferior, con perforación nasal y hernia del iris; paño corneal; disminución de la profundidad de la cámara anterior, con leve celularidad y opacidad lenticular (fig. 1).
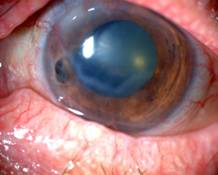
Al cabo de los días comenzó el mismo problema en el ojo derecho, que presentó reacción ocular moderada, sensación de cuerpo extraño, lagrimeo y disminución de la visión.
En los anexos se observó hiperemia conjuntival bulbar, edema de párpados con costras amarrillas, erosión, vesículas en las conjuntivas palpebrales y secreción conjuntival amarilla. De igual modo, en la biomicroscopia con lámpara de hendidura del segmento anterior se observó infiltrado corneal central extenso, perforación pequeña con pigmentos de iris coincidentes con las horas 5-6 del reloj, disminución de la profundidad de la cámara anterior, con leve actividad inflamatoria y opacidad lenticular (fig. 2).
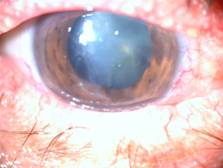
Tratamiento aplicado
Respecto al tratamiento sistémico, el paciente consumía prednisona de 80 mg y azatioprina de 100 mg.
El tratamiento local se comenzó con colirios fortificados de antibióticos (ceftazidima y amikacina), midriático, antiinflamatorio no esteroideo, agentes antihipertensivos y oclusiones.
Se realizaron estudios microbiológicos donde se aisló el Staphylococcus aureus. Luego de lograr controlar la infección, se colocó un lente blando de contacto con fin terapéutico y se reajustó la dosis medicamentosa.
Al cabo de algunas semanas se obtuvo cicatrización total del ojo derecho y parcial del ojo izquierdo, por lo que se decidió retirar el lente del ojo derecho e iniciar la aplicación de lubricante oftalmológico sin preservo, mientras que en el ojo izquierdo se mantuvo el tratamiento hasta que se logró que el epitelio cubriera el estroma del iris, se formara la cámara anterior y desapareciera el proceso inflamatorio; también se prescribieron lubricantes y se cambió el lente blando. Existió mejoría de la agudeza visual en ambos ojos.
El tratamiento sistémico se continuó bajo control estricto de los dermatólogos. En la actualidad el paciente presenta estabilidad ocular y continúa siendo atendido en la consulta de Oftalmología.
Comentarios
El pénfigo vulgar fue distinguido del resto de las enfermedades dermatoampollosas por Walter Lever, en 1953. El término proviene del griego "pemphix" o "akantha " y con él se denomina a un grupo de enfermedades autoinmunes que producen vesículas y ampollas cutáneas y de las membranas mucosas por la acción de autoanticuerpos contra proteínas específicas localizadas en las uniones de las células del epitelio.4 Las ampollas son consecuencia de una pérdida de la adhesividad entre las células epidérmicas -proceso conocido como acantólisis-, lo cual se cree que pueda ser causado por un proceso autoinmune. Esta enfermedad es poco frecuente, de evolución maligna y crónica, con inicios impredecibles, por lo que la persona afectada requiere tratamiento continuo y sistemático, a fin de evitar un desenlace letal.
Se caracteriza por la presencia de anticuerpos de inmunoglobulina G circulantes contra la desmogleína 3 (Dsg3) en la sustancia intercelular de los epitelios. Alrededor de la mitad de los pacientes también posee anticuerpos contra la Dsg1. Existe una cierta susceptibilidad mayor ante la presencia de factores de genes del HLA de clase I (HLA-A10, HLA-A26) y sobre todo a la asociación con moléculas de HLA de clase II (DR4, DR14).7
Si bien en ocasiones son atendidos pacientes en edades juveniles, el PV aparece con mayor frecuencia a partir de la cuarta y hasta la sexta décadas de la vida, sin diferencias en cuanto a raza o género (afecta a ambos sexos por igual).
En más de 88 % de los pacientes, el PV se manifiesta con lesiones bucales y pueden pasar meses antes de que la enfermedad aparezca en la piel. La lesión elemental son las ampollas, que suelen ser múltiples, mal definidas, de distinto tamaño y cubierta fina, las que se rompen fácilmente produciendo erosiones superficiales, irregulares y muy dolorosas. Otros signos concomitantes son la formación de nuevas ampollas, junto con otras ya evolucionadas, y de úlceras, lo que indica un carácter progresivo. Así, se pueden observar ampollas íntegras, ampollas en las que la superficie se está desprendiendo y aparece como una auténtica membrana de tejido organizado, que se puede separar con una sonda; o pseudomembranas, que cubren las erosiones formadas fundamentalmente por un exudado inflamatorio, que no constituye un tejido organizado que pueda separarse con un explorador.7
Las manifestaciones oculares son subestimadas en su frecuencia por el poco reconocimiento de los signos característicos. Las características clínicas del pénfigo ocular indican a la conjuntivitis y la blefaritis como las entidades más comúnmente manifestadas, con síntomas de irritación ocular, lagrimeo y sensación de cuerpo extraño. Solo en la minoría de los casos se presentan vesículas y erosiones en la conjuntiva bulbar y la palpebral; sin embargo, la agudeza visual no se ve afectada. En personas con conjuntivitis cicatrizante crónica de causa dudosa, es útil la biopsia de conjuntiva para confirmar el diagnóstico de PV.
La presencia de daño corneal es rara, pero cuando se ha notificado la afectación, resulta grave, con erosiones corneales que evolucionan a la perforación ocular, a pesar del tratamiento sistémico invasivo con esteroides e inmunosupresores. En los informes de casos se refiere el proceso infeccioso como la causa más común de la erosión, puesto que la terapia con inmunosupresores y esteroides puede afectar la superficie ocular y predisponer a la infección.4
En el paciente de este caso clínico existían signos de infiltración estromal y celularidad en la cámara anterior, así mismo se confirmó la infección por un microorganismo patógeno, que se indicó como causa del proceso morboso.

