










 Serviços customizados
Serviços customizados Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroducción
Los electrodos selectivos a iones (ESIs) son un subgrupo importante de sensores electroquímicos que se caracterizan por su pequeño tamaño, portabilidad, alta selectividad, sencillez, bajo consumo de energía y bajo costo, características atractivas para su aplicación en análisis químico.1,3,4 La selectividad inherente de estos dispositivos se atribuye a interacciones altamente selectivas entre el material de la membrana y el ion objetivo.
La interferencia ocurre cuando una sustancia o proceso conduce a un resultado falso en una técnica de medición.5 A menudo, solo es de interés la selectividad de un sensor con una composición de membrana determinada, que se puede caracterizar con un coeficiente de selectividad termodinámicamente fundado para cada ion interferente.6 Este coeficiente de selectividad ( ) define la capacidad del ESI para distinguir el ion de interés de los iones interferentes y está definido por la ecuación 1:
) define la capacidad del ESI para distinguir el ion de interés de los iones interferentes y está definido por la ecuación 1:
 (1)
(1)donde:
es el coeficiente de selectividad,
 es la diferencia de los potenciales individuales extrapolados a la actividad de 1M para los iones
es la diferencia de los potenciales individuales extrapolados a la actividad de 1M para los iones 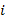 y
y  , z es el número de electrones,
, z es el número de electrones,
R la constante de los gases,
F la constante de Faraday
T la temperatura absoluta.
O sea, el coeficiente de selectividad es una función directa de las diferencias de los potenciales individuales extrapolados a la actividad de 1M para los iones y
, es decir, el desplazamiento vertical entre las curvas de calibración individuales en actividades de muestra de 1M. Los valores correctos de se obtienen a partir de medidas adecuadas e insesgadas de  para cada ion. Los coeficientes de selectividad se pueden usar convenientemente para predecir funciones de respuesta en muestras mixtas.5
para cada ion. Los coeficientes de selectividad se pueden usar convenientemente para predecir funciones de respuesta en muestras mixtas.5
Los procedimientos tradicionales para evaluar los valores de son el método de solución separada 7 y el método de interferencia fija.8 Por supuesto, si los valores de están sesgados, sus valores numéricos dependerán de las condiciones experimentales.
En el caso de los electrodos de membrana polimérica, las interferencias de otros iones en la muestra vienen dictadas principalmente por su extracción competitiva en la fase orgánica. En consecuencia, la respuesta de estos ESIs suele ser completamente predecible a partir de constantes termodinámicas como, en el caso de los sistemas basados en ionóforos, la constante de formación de cada complejo ion-ionóforo en la membrana. Dado que las selectividades de los electrodos de membrana polimérica están directamente relacionadas con los equilibrios de las dos fases, se establece un puente importante con la química de anfitrión-huésped.6
Es de esperar entonces, que la secuencia de constantes de estabilidad para complejos de varios iones metálicos dependa de los radios iónicos y electronegatividades de los respectivos iones metálicos. Las explicaciones de los factores que determinan la especificidad de la absorción de iones metálicos también se basan a menudo en principios cuantitativos o semicuantitativos, como el principio HSAB (ácidos y bases duros y blandos) de Parr y Pearson, 9,10 la serie de Irving-Williams de constantes de estabilidad 11,12 y la abundancia de geometrías de coordinación de metales de transición en las estructuras moleculares determinadas experimentalmente.13,14
Es un interés de la investigación teórica, calcular parámetros que puedan correlacionarse con información experimental, en este caso, los coeficientes de selectividad potenciométricos de los electrodos selectivos a iones. Ello resulta un reto debido a la diversidad de factores que intervienen. De lograrse correlaciones aceptables se podrían diseñar ionóforos “a la medida” con menores interferencias que oriente a los experimentalistsa en la construcción de los ESIs apropiados.
En la actualidad, se emplean diferentes procedimientos para estimar los coeficientes de selectividad potenciométricos tales como: Teoría del Funcional de la Densidad (DFT), estudios de relación cuantitativa estructura-propiedad (QSPR), tanto con redes neuronales artificiales (ANN) 15,16, como con máquinas de soporte vectorial (VSM) 17 o con algoritmo genético (GA).18) Como puede apreciarse la química cuántica ha abordado poco el tema, y cuando lo ha hecho, ha usado la Teoría de los Funcionales de la Densidad.
Existen divergencias en cuanto a las justificaciones teóricas que tratan de explicar la selectividad en un electrodo selectivo a iones. La mayoría de los enfoques teóricos de los ESIs basados en ionóforos neutros interpretan la selectividad potenciométrica en términos de parámetros de equilibrio termodinámico. Según este grupo de teorías, el coeficiente de selectividad es proporcional a la relación de las constantes de extracción. También se conoce otro enfoque de la selectividad: el enfoque cinético. Este enfoque alternativo asume un estado estable en la interfaz membrana/solución. Por lo tanto, en lugar del potencial de equilibrio, se establece un potencial mixto y los coeficientes de selectividad son iguales a las relaciones de las densidades de corriente de intercambio para los iones interferentes y primarios.19
Lo que queda claro de ambas teorías es que la selectividad potenciométrica depende de los fenómenos químico-físicos de la interfase membrana/solución. O sea, depende, entre otros factores, de la composición de la membrana, la que a su vez depende, además del ionóforo usado, de la matriz y de los disolventes mediadores o plastificantes, e incluso varía con el tiempo de vida del electrodo y por lo tanto también el coeficiente de selectividad potenciométrico.20 Por lo tanto, aunque existe una relación estrecha entre la estabilidad de la unión ion-ionóforo y el coeficiente de selectividad potenciométrico, esta estará influenciada por otros factores.
La N,N'-((1,3-fenilenbis(azanediil))bis(carbonotioil))bis(furan-3-carboxamida) (FTB), al igual que otras tioureas y bistioureas han sido empleadas en la construcción de ESIs a diferentes iones metálicos como Cu2+, Pb2+ y Cd2+.21,22,23,24,25,26) En el caso particular de la FTB se encuentra reportada como ionóforo en la construcción de un ESI a iones Pb2+ con membrana de PVC.26 En dicha investigación se calcularon los coeficientes de selectividad potenciómetros mediante el método de la interferencia fija, sin embargo no se profundiza en la justificación teórica de estos resultados.
Cuando los logaritmos decimales de los coeficientes selectividad potenciométricos son valores superiores a -2, se considera que el ion es un fuerte interferente al ion para el que se hizo el electrodo selectivo. Así en el trabajo experimental de obtención del electrodo selectivo a Pb2+ con FTB como ionóforo, Wilson, D. y colaboradores 26 reportan que el Cu2+ es fuerte interferente en tanto el resto de los iones no tienen significativas interferencias desde el punto de vista analítico, al menos cuando su concentración es comparable con la concentración de Pb2+, lo que se muestra en la tabla 1.
Tabla 1 Logaritmo decimal de los coeficientes de selectividad potenciométrico de los principales iones interferentes.
| Interferencias | Cu2+ | Cd2+ | Zn2+ | Hg2+ | Co2+ | Ni2+ | Ca2+ |
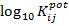
|
-1,0 | -2,1 | -2,7 | -3,0 | -3,4 | -3,2 | -3,7 |
El objetivo de la investigación es proponer un nivel de teoría y una estrategia de cálculo que pueda ser capaz de predecir con un margen aceptable, las principales interferencias que ofrecen iones de igual carga a la aplicación de un ESI a Pb2+ construido con el ionóforos FTB y que oriente a explorar su aplicación a sistemas análogos.
Materiales y métodos
La estrategia general de cálculo para lograr este objetivo se resume en determinar las energías de unión entre la FTB y cada ion metálico: Pb2+, Cu2+, Hg2+, Zn2+, Ni2+, Co2+, Ca2+ y Cd2+, aplicando DFT a tres niveles de bases de datos y considerando la interacción directa ionóforo-ion y ionóforo-ion solvatado con agua para correlacionar los resultados con los coeficientes de selectividad potenciométricos.
Todos los cálculos donde se optimizan las estructuras se realizaron a condiciones estándar de temperatura (298K) utilizando el software GAMESS. Se comprobó que se había optimizado a un mínimo en la Superficie de Energía Potencial por los valores positivos de todas las frecuencias vibracionales.
Selección de las Geometrías de coordinación y multiplicidad de spin
Las geometrías de coordinación preferidas de cada catión metálico han sido estudiadas y hoy en día son perfectamente conocidas. En cuanto a las geometrías más comunes, la tetraédrica es preferida por el Zn2+, la cuadrado planar por el Cu2+, la lineal por el Hg2+ y la octaédrica por Co2+, Ni2+, pero también pueden asumir geometrías tetraédricas. El Cd2+ tiene geometrías tanto octaédrica como tetraédrica.14 Los iones Ca2+ se pueden encontrar formando complejos con geometría de antiprisma cuadrado.13
La geometría alrededor de Pb2+ es diversa, así la literatura reporta al menos cinco poliedros de coordinación diferentes, donde los números de coordinación varían entre 2 y 10.27 La diversidad de geometrías de coordinación depende de si el par de electrones sin compartir es estereoquímicamente activo o inactivo. Estos dos casos se denominan “holodirectos”, en los que los enlaces a los átomos del ligando se dirigen a lo largo de la superficie de un globo circundante y "hemidirectos”, en los que los enlaces a los átomos del ligando se dirigen sólo a una parte del globo, es decir, hay un vacío (o brecha) identificable en la distribución de enlaces a los ligandos.28) Los compuestos de Pb2+ están hemidireccionados para números de coordinación bajos (2-5) 29) y holodireccionados para números de coordinación altos (9 y 10) 30, en tanto para números de coordinación intermedios (6-8), se han informado ejemplos de ambos tipos de estereoquímica.31,32
En esta investigación, de acuerdo con las posibilidades del ionóforo, se asume que la relación ion- ionóforo más probable es 1:1, por lo que se calculan solo las geometrías que no exceden el número de coordinación 4, o sea, lineales, tetraédricas y cuadrado planares.
En cuanto a la multiplicidad de spin en el caso de los iones Zn2+, Cd2+ y Hg2+ todos presentan distribución d10 y Ca2+, p6, por lo que son sistemas de capa cerrada con singlete como único estado base posible. Los iones Cu2+ presentan distribución d9 con doblete como único estado base posible. Sin embargo, en el caso de los iones Co2+ y Ni2+ pueden existir en sus complejos tanto en estados de alto como de bajo spin. El ion Ni2+ presenta distribución d8 por lo cual puede tener estados de triplete y singlete, pero es conocida su preferencia por formar complejos de bajo spin y el Co2+ con distribución d7 puede tener estados de cuadruplete y doblete, pero forma normalmente complejos de alto spin.33-34
Selección del nivel de teoría del cálculo
Para investigar la afinidad inherente del ionóforo FTB por varios iones metálicos, se utilizaron cálculos teóricos para determinar las estructuras y las energías de unión para los complejos metálicos con FTB (FTB-Mx+) para M = Pb2+, Cu2+, Hg2+, Zn2+, Ni2+, Co2+, Ca2+ y Cd2+ , el ionóforo FTB y los iones metálicos seleccionados.
Las estructuras fueron optimizadas con el método DFT, eligiéndose el funcional M06L y tres sistemas de bases de funciones.
La selección del funcional M06L se hizo porque, al igual que toda la familia de funcionales de Minnesota, se ha vuelto popular para el caso de la química de elementos de transición, en tanto fue parametrizado para tener un buen rendimiento tanto en sistemas con metales representativos como de transición. El funcional M06L se ha empleado satisfactoriamente para sistemas con metales de transición en estudios de termoquímica, cinética termoquímica, organometálica, inorgánica, interacciones biológicas e interacciones no covalentes. Está recomendado para simulaciones en sistemas con tamaños moderados y grandes y cuando se deben abordar escalas de tiempo largas.35
Se seleccionaron tres conjuntos de base de funciones. El primero combina la base 6-31G(d) para los átomos C, O, H, S y N y la base SDD para los metálicos; el segundo combina la base 6-31+G(d,p) para los átomos C, O, H, S y N con SDD+f para los metálicos y el tercero las bases 6-31++G(3df,3pd) para los átomos C, O, H, S y def2-QZVPPD para los átomos metálicos. Estos tres niveles de cálculo permitirán valorar el efecto que tienen las bases de funciones en los resultados energéticos al contrastarlos con los índices de selectividad experimentales.
Cálculo de las energías de unión, hidratación y relativas
La energía de unión es la cantidad de energía que se libera cuando se crea un sistema vinculado. Es lo que mantiene unido al sistema y mientras mayor sea, mayor será su estabilidad. Por lo tanto, la energía de unión se puede relacionar proporcionalmente con la afinidad que tiene un determinado compuesto para unirse a otro.
La energía de unión se calcula mediante la diferencia de la energía del complejo con la suma de las energías de sus componentes por separado, calculadas al mismo nivel de teoría (ecuación 2).
 (2)
(2)donde
Eb es la energía de unión ion-ionóforo,
EM-L es la energía de geometría optimizada de la unión ion-ionóforo en el vacío,
EM, la energía del ion en fase gaseosa y
EL la energía optimizada del ionóforo libre.
El efecto de la solvatación de los iones en el medio acuoso donde ocurre la determinación experimental, se modeló calculando la energía de hidratación como (ecuación 3):
 (3)
(3)donde:
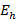 es la energía de hidratación para cada catión,
es la energía de hidratación para cada catión,
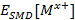 es la energía calculada aplicando el modelo de solvatación SMD y
es la energía calculada aplicando el modelo de solvatación SMD y
 es la energía del catión en fase gaseosa.
es la energía del catión en fase gaseosa.
El cálculo de la energía de solvatación se realizó a los mismos niveles de teoría en que se hizo para el sistema en el vacío, al que se le incorporó el modelo de solvatación SMD, atendiendo a que el mismo está parametrizado para la familia de funcionales de Minnesota.36
Como el propósito de la investigación es modelar los posibles interferentes, resulta más pertinente calcular la energía relativa (Er) de cada interferente con respecto al ion para el que se construyó el ESI (Pb2+), para lo que se toma como base la reacción química (ecuación 4):
 (4)
(4)Que indica la competencia entre el ion 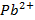 hidratado (o en general el ion para el que se construye el electrodo selectivo) con el otro ion hidratado por formar el ion-complejo. Es expresada en la ecuación que define la energía relativa del posible interferente con el ion al que es selectivo el ESI (ecuación 5):
hidratado (o en general el ion para el que se construye el electrodo selectivo) con el otro ion hidratado por formar el ion-complejo. Es expresada en la ecuación que define la energía relativa del posible interferente con el ion al que es selectivo el ESI (ecuación 5):
 (5)
(5)donde
Eh[Pb2+] y Eh[Mx+] son las energías de hidratación del Pb2+ y de cada ion interferente y EB[FTB-Pb2+] y EB[FTB-Mx+] son las energías de unión ion-ionóforo para el Pb2+ y cada ion interferente.
El cambio de energía para esta reacción puede usarse como una indicación de la selectividad de FTB por Pb2+ en comparación con Mx+. Un valor negativo de Er indica selectividad de FTB para Pb2+ sobre Mx+, mientras que un Er positivo indica selectividad para Mx+ sobre Pb2+. Mientras más cercano a cero sean los valores negativos de Er la afinidad del ion interferente por FTB tendrá más semejanza a la del Pb2+.
Resultados y discusión
Los datos geométricos más significativos del ionóforo se muestran en la tabla 2 y la identificación de los átomos se observan en la figura 1.

Tabla 2 Selección de distancias de enlace (en Å), ángulos y ángulos diedros (en grado) más significativos del ionóforo FTB optimizadas con el funcional M06L y tres bases de funciones.
| Distancias | 6-31G(d) | 6-31+G(d,p) | 6-31++G(3df,3pd) |
| C16-S18 | 1,638 4 | 1,636 8 | 1,630 9 |
| C15-S17 | 1,638 4 | 1,636 8 | 1,630 9 |
| C24-O26 | 1,211 4 | 1,212 9 | 1,208 6 |
| C23-O25 | 1,211 4 | 1,212 9 | 1,208 6 |
| Ángulos | |||
| N13-C16-S18 | 126,60 | 126,60 | 126,42 |
| N11-C15-S17 | 126,60 | 126,60 | 126,42 |
| N21-C24-O26 | 124,51 | 124,35 | 123,60 |
| N19-C23-O25 | 124,51 | 124,35 | 123,60 |
| Ángulos diedros | |||
| C4-N13-C16-S18 | 5,69 | 6,46 | 7,77 |
| C6-N11-C15-S17 | -5,68 | -6,46 | -7,77 |
| C24-N21-C24-O26 | 18,09 | 19,46 | 14,33 |
| C15-N19-C23-O25 | -18,09 | -19,46 | -14,33 |
| O26-C24-C27-O29 | 13,19 | 13,08 | 12,03 |
| O25-C23-C35-O37 | -13,19 | -13,08 | -12,03 |
Como se puede apreciar la geometría del ionóforo FTB es semejante sin importar el conjunto de base elegido y donde las cadenas que se unen al anillo bencénico se encuentran bastante coplanares, Las geometrías optimizadas pertenecen al grupo puntual de simetría Cs. Esta simetría propicia que los átomos de oxigeno O25, O26 se encuentren muy separados como se aprecia por simple inspección en la figura 1, por lo que se puede esperar que la molécula sufra deformaciones a la hora de formar los complejos ion-ionóforo tetradentados. En cuanto a la disposición de los azufres S17 y S18 parecen propicios para la formación de enlaces coordinados sin que la molécula sufra grandes deformaciones geométricas en sus entornos.

Fig. 2 Geometrías optimizadas de los complejos FTB-Mx+ a M06L/6-31++G(3df,3pd), def2-QZVPPD. (a) M=Ni2+, (b) M=Zn2+, (c) M=Hg2+, (d) M=Pb2+, (e) M=Ca2+, (f) M=Cd2+, (g) M=Co2+, (h) M=Cu2+
Los datos de las geometrías más significativas de los complejos, relacionadas con el entorno al ion metal-ionóforo se muestran en la tabla 3 y su representación gráfica optimizados al más alto nivel de teoría M06L/6-31++G(3df,3pd), def2-QZVPPD se aprecian en la figura 2. De su análisis se identifica que el ionóforo FTB se comporta como tetradentado para todos los complejos ion-ionóforo, excepto para el Hg2+ que se comporta como bidentado y donde las distancias Hg2+- O26 y Hg2+- O27 son superiores a 5.151 A, lo cual es algo esperado, ya que este ion generalmente se presenta formando geometrías lineales. Las geometrías de los complejos tetradentados no coinciden en su totalidad con un cuadrado plano ni con un tetraedro, sino que asumen geometrías deformadas, lo que puede apreciarse en los ángulos diedros reportados.
Tabla 3 Selección de distancias (en Å) del ion metálico con los heteroátomos y ángulos diedros (en grado) más significativos de los complejos FTB-Mx+, calculadas al nivel más alto de teoría M06L/6-31++G(3df,3pd), def2-QZVPPD
| Metal/Enlace | S18-M27 | S17-M27 | O26-M27 | O25-M27 |
| Ca2+ | 2,786 12 | 2,859 42 | 2,218 90 | 2,203 12 |
| Cd2+ | 2,605 04 | 2,604 11 | 2,214 53 | 2,241 52 |
| Co2+ | 2,417 3 | 2,411 4 | 1,946 0 | 1,956 7 |
| Cu2+ | 2,356 5 | 2,478 1 | 1,914 4 | 1,910 5 |
| Ni2+ | 2,258 6 | 2,378 2 | 1,850 0 | 1,840 0 |
| Hg2+ | 2,471 0 | 2,471 0 | 5,151 4 | 5,151 5 |
| Pb2+ | 2,795 5 | 2,947 9 | 2,523 3 | 2,366 5 |
| Zn2+ | 2,388 0 | 2,407 1 | 1,972 2 | 2,004 1 |
| Metal/Diedros | C4-N13-C16-S18 | C6-N11-C15-S17 | C24-N21-C24-O26 | C15-N19-C23-O25 |
| Ca2+ | -24,17 | -115,19 | 17,02 | 24,73 |
| Cd2+ | -30,57 | -117,34 | 22,41 | 29,08 |
| Co2+ | -31,22 | -110,48 | 19,67 | 22,22 |
| Cu2+ | -40,31 | -111,33 | 18,29 | 23,82 |
| Ni2+ | -50,63 | -115,50 | 27,65 | 27,65 |
| Hg2+ | 10,29 | -10,18 | 12,25 | -12,27 |
| Pb2+ | -24,91 | -120,24 | 21,44 | 27,42 |
| Zn2+ | -31,97 | -107,95 | 21,91 | 25,95 |
Los resultados del cálculo de las energías de unión ion-FTB obtenidas de las energías de las geometrías optimizadas a los tres niveles de teoría se muestran en la tabla 4.
Tabla 4 Energías de unión en kilocaloría/mol calculadas a M06L para cada conjunto de base de los complejos (M-FTB)x+.
| Conjunto de base/ Mx+ | Ca2+ | Cd2+ | Co2+ | Cu2+ | Ni2+ | Pb2+ | Zn2+ | Hg2+ |
| 6-31G(d), SDD | -210,25 | -274,48 | -346,39 | -374,26 | -421,47 | -202,48 | -337,84 | -316,85 |
| 6-31+G(d,p), SDD+f | -207,06 | -269,09 | -341,2 | -367,3 | -415,48 | -202,74 | -331,81 | -312,73 |
| 6-31++G(3df,3pd), def2-QZVPPD | 126,68 | 69,87 | 13,73 | -21,72 | -22,75 | 121,97 | 18,01 | 30,04 |
Se aprecia que las energías de unión más elevadas corresponden a los complejos con los cationes Ni2+, Cu2+ y Co2+, por ese orden, independientemente de la calidad de las bases empleadas y sorprendentemente el catión del complejo con Pb2+ es siempre el de menor valor. Este resultado es indicativo de que empleando el funcional de densidad M06L, independientemente de cuánto se mejore la calidad de la base de funciones, los resultados no se pueden correlacionar adecuadamente con las constantes de afinidad por el ionóforo reportadas en el experimento (tabla 1) si no se incorpora al cálculo teórico el efecto de la solvatación.37
Los valores calculados de la energía de hidratación y la energía relativa se presentan en las tablas 5 y 6 respectivamente. De acuerdo a los resultados experimentales se debe esperar que todos los valores de energía relativa sean exotérmicos, lo cual indicaría la selectividad hacia el Pb2+ con respecto a otros metales, y que el menos exotérmico sea el del Cu2+, pero para los cálculos a M06L y conjuntos de base 6-31G(d), SDD+f y 6-31+G(d,p), SDD+f no ocurre de esta manera. Sin embargo, para el conjunto de base 6-31++G(3df,3pd), def2-QZVPPD sí aprecia concordancia con los resultados experimentales. Incluso se puede observar cómo el valor del Cu2+ es prácticamente similar al Pb2+, lo cual concuerda con su comportamiento como alto interferente en la determinación de Pb2+. Por otro lado, la baja energía de hidratación del Pb2+ compensa sus valores de energía de unión, lo cual es otro indicativo de la necesidad de incluir la solvatación en la estimación.
Tabla 5 Energías de hidratación en kcal/mol calculadas a M06L y modelo de solvatación SMD para cada conjunto de base.
| Conjunto de base/Mx+ | Ca2+ | Cd2+ | Co2+ | Cu2+ | Ni2+ | Pb2+ | Zn2+ | Hg2+ |
| SDD | -386,36 | -419,49 | -463,03 | -473,80 | -404,10 | -329,28 | -475,90 | -434,26 |
| SDD+f | -386,37 | -419,49 | -463,03 | -473,89 | -402,59 | -329,28 | -475,92 | -434,25 |
| def2-QZVPPD | -386,37 | -418,94 | -461,96 | -473,32 | -403,91 | -326,47 | -475,89 | -433,14 |
Tabla 6 Energías relativas en kcal/mol calculadas para complejos (M-FTB)x+ a M06L con cada conjunto de base.
| Conjunto de base/ Mx+ | Ca2+ | Cd2+ | Co2+ | Cu2+ | Ni2+ | Pb2+ | Zn2+ | Hg2+ |
| 6-31G(d), SDD | -49,32 | -18,22 | 10,16 | 27,26 | 144,16 | 0,00 | -11,27 | 9,39 |
| 6-31+G(d,p), SDD+f | -52,77 | -23,86 | 4,71 | 19,96 | 139,44 | 0,00 | -17,57 | 5,03 |
| 631++G(3df,3d), def2-QZVPPD | -64,60 | -40,36 | -27,24 | -3,14 | 67,30 | 0,00 | -45,45 | -14,73 |
La diferencia entre los resultados para cada combinación de conjunto de base está justificada teniendo en cuanta la calidad del conjunto de base. Para el ionóforo FTB, el conjunto de base 6-31++G(3df,3pd) emplea más polarizaciones y funciones difusas que 6-31G(d) y 6-31+G(d,p), lo cual le permite mucha más flexibilidad y con ello calcular mejor este tipo de interacciones que implican la deformación de la nube electrónica de los átomos involucrados en la formación de los enlaces coordinados. De igual forma ocurre con los iones metálicos. El conjunto de base def2-QZVPPD es mucho más extenso que el SDD y el SDD+f, e incluye un potencial de núcleo efectivo (ECP) mucho más extenso, además de una mayor polarización para el metal.
El diseño del cálculo llegando hasta una base tan poderosa como la 6-31++G(3df,3pd), nos permitió encontrar un nivel de teoría que fuera capaz de reproducir el patrón de interferencia experimental de iones bivalentes sobre el ion selectivo del ESI construido. Ello significó un costo computacional no despreciable en comparación con las bases menos extensas empleadas en el estudio, pero siempre resulta en una ganancia de recursos y tiempo si se usara con fines predictivos. Para el tamaño de los sistemas estudiados, la combinación 6-31++G(3df,3pd), def2-QZVPPD no implica tampoco tiempos de cálculo excesivos.
Como defecto del modelo de abordar el estudio, cabe señalar, que los cálculos fallan a la hora de estimar la afinidad del ionóforo por el ion Ni2+. Incluso para la combinación 6-31++G(3df,3pd) y def2-QZVPPD este ion presenta valores endotérmicos de energía relativa, lo cual no significa que el procedimiento de cálculo es fallido en su totalidad, ni que no se pueda estimar los coeficientes de selectividad con este procedimiento. A sabiendas de las limitaciones de simular una magnitud tan compleja era de esperar que no se obtuviera un modelo totalmente exacto, ni que se pudiera alcanzar una relación cuantitativa entre las energías relativas y los coeficientes de selectividad.
La complejidad de abordar el tema se muestra en los resultados obtenidos por S. Demir y colaboradores, quien para un ionóforo selectivo a Cu2+ que muestra como interferentes principales al Hg2+ y Cr3+ solo puede demostrar la interferencia del Cr3+, pero no del Hg2+ mientras son sobreestimados los iones Co2+ y Ni2+ de forma análoga a esta investigación.38
En comparación con las teorías que normalmente se usan para predecir la selectividad, como la teoría de ácidos y bases duros y blandos de Pearson (HSAB), concuerdan en la similitud en la afinidad entre el Cu2+ y el Pb2+, que son clasificados como ácidos de dureza intermedia. Sin embargo de acuerdo a la HSAB los cationes Hg2+ y Cd2+, que se clasifican como ácidos blandos, tienden a formar complejos muy estables con los compuestos que contengan azufre, y deberían ser también fuertes interferentes, o incluso, más afines que el Pb2+, lo cual no está de acuerdo con los resultados experimentales, ni con los calculados por DFT.
Conclusiones
Las energías relativas de la interacción ente el ionóforo FTB y los iones metálicos solvatados, calculadas al nivel de teoría M06L/6-31++G(3df,3pd), de f2-QZVPPD reproducen esencialmente el comportamiento de las interferencias deducidas de los coeficientes de selectividad potenciométricos reportadas experimentalmente para siete iones, excepto para los iones Ni2+. El uso de bases de menores niveles de polarización y menos difusas, aun cuando se considere la hidratación de los iones, no es suficiente para estimar de forma aceptable los resultados experimentales del sistema estudiado.

