










 Serviços customizados
Serviços customizados Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
El síndrome de Townes-Brocks (TBS) fue descrito en 1972, está constituido por un cuadro malformativo multisistémico que presenta herencia autosómica dominante. Los defectos que se observan en los pacientes muestran un patrón de manifestación variable, pero la mayoría tienen dos o más de las siguientes anomalías: alteraciones auriculares y de oído, como los apéndices preauriculares, microtia; defectos de extremidades, como pulgares bífidos o trifalángicos, hipoplasia de pulgares, defectos radiales, ausencia de dedos y sindactília; atresia o estenosis en el ano, con o sin fístula; también se recogen antecedentes familiares con el mismo defecto.1,2,3
Sin embargo, otros órganos pueden también estar afectados, dentro de los que se han descrito algunos casos que presentaban defectos del tabique ventricular, hipoplasia renal, anomalías craneofaciales semejantes al complejo de Goldenhar, y en algunos casos deficiencia mental. Aunque la mayoría de los casos descritos son familiares, también se presenta de forma esporádico; se reportan en diversas poblaciones que incluyen los Estados Unidos, Brazil, Francia, España, Portugal Alemania, Inglaterra y Rusia. Investigaciones anteriores mostraron la gran variabilidad en la expresividad del síndrome, tanto inter como intrafamiliar; resultando importante el interrogatorio a los familiares y examen físicos a los progenitores.4,5
En 1998, se identificó que esta enfermedad está causada por una mutación en el gen específico del factor de transcripción SALL1 localizado en el cromosoma 16q12.1.1,6
La expresividad variable de este síndrome hace difícil el diagnóstico clínico, por lo que resulta muy importante el examen físico exhaustivo para evaluar los signos dismorfológicos malformativos que permite realizar un diagnóstico correcto. Con el objetivo de describir estos signos clínicos se realizar esta presentación.
PRESENTACIÓN DE CASO
Lactante femenina de nueve meses operada al nacimiento por ano imperforado a la que se le realizó colostomía, acude a consulta para evaluar su desarrollo psicomotor, así como su estado nutricional.
Antecedentes patológicos familiares: no refieren ninguno de interés.
Antecedentes prenatales: estudios prenatales bioquímicos y ultrasonográficos normales.
Antecedentes perinatales: nacida por un parto eutócico a las 39,5 semanas, con peso 2 980 gramos, talla 49,5 cm; apgar 8-9, al examen físico el neonatólogo observa apéndices preauriculares y orejas displásicas, primer artejo de la mano derecha bífido, región del periné corto con un pliegue profundo que sugiere ano anterior que no está permeable; se efectuó de inmediato el diagnóstico de ano imperforado, se valoró por cirugía neonatal y se le realizó colostomía.
Se valora nuevamente en consulta de Genética Clínica a lactante y se aprecia buena ganancia de peso; buen desarrollo psicomotor y pondoestatural. Se realizó evaluación peso/edad: 8 250 gramos y talla/edad: 69cm, entre 25-50 percentiles. La valoración ponderal fue adecuada para su edad y sexo.
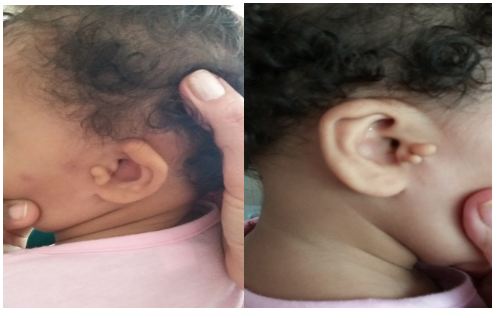
Al examen físico se observan en varios apéndices preauriculares en ambas orejas con displasia de estas y sobre plegamiento del hélix, más acentuado en la oreja izquierda. (Fig. 1)
La malformación de los artejos con preferencia del primer dedo, en este caso el pulgar bífido de la mano derecha. (Fig. 2)

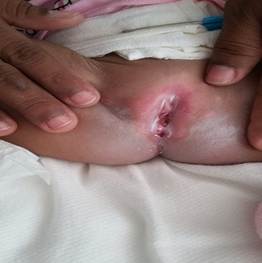
En la región del periné se observan pliegues en región anterior que sugieren un ano que no es permeable. (Fig. 3)
Se solicitó a la familia consentimiento informado para examinarla y tomarle fotografías de los defectos para ser publicadas en revistas científicas.
DISCUSIÓN
El síndrome Townes-Brocks lo forman anomalías congénitas, con una frecuencia de un enfermo cada 350 000 nacidos vivos. Los afectados al nacimiento exhiben una triada malformativa de ano imperforado (84 %), orejas displásicas (87 %), con hélices superiores sobreplegados y apéndices preauriculares; frecuentemente asociados a discapacidad auditiva neurosensorial o conductiva (65 %) y las malformaciones del pulgar (89 %); polidactilia preaxial, pulgares trifalángico o bífido o anchos también hipoplásico o ausentes). Pudieran presentarse otras alteraciones anorrectales (atresia anal con fístula o no, ano anterior y estenosis anal) y en menor frecuencia alteraciones renales (42 %), cardíacas y genitales. 1,3,5
El TBS se hereda de forma autosómica dominante, con alta penetrancia y gran heterogeneidad clínica. Es causada con alta frecuencia por mutaciones patogénicas nuevas o de novo, estimadas en un 50 %. Cada hijo de un individuo enfermo con una mutación en el gen SALL1 tiene un 50 % de posibilidades de heredar este síndrome polimalformativo. El diagnóstico prenatal de los embarazos con mayor riesgo es posible si la mutación ha sido identificada en la familia.1,2,6
Las anomalías en el oído externo varían desde un hélix superior replegado, antehélix pequeño o, típicamente, presencia de apéndices preauriculares. Un hallazgo común en estos niños es la hipoacusia primaria, sobre todo neurosensorial, a veces progresiva, y más acentuada en frecuencias altas. Sin embargo, en algunos pacientes la hipoacusia también tiene un componente de transmisión, sobre todo si existen alteraciones en oído medio, tales como hipoplasia de cabeza de martillo o anomalías en la ventana oval.3,7
En la región anorrectal, la alteración más frecuentemente registrada es la imperforación anal, más o menos alta, acompañada a veces de fístulas rectovaginales o rectouretrales.2,8
En las extremidades, el hallazgo más frecuente a este nivel es el pulgar trifalángico y la polidactilia preaxial, aunque también son comunes otras alteraciones como la sindactilia, desviación cubital de pulgar o pulgar bífido. Las malformaciones a nivel de miembros inferiores son menos habituales (se han encontrado pacientes con superposición de 2º y 4º dedos sobre el 3º, sindactília de 3º y 4º dedos).6,7
Las anomalías genitourinarias, con una alta incidencia consistentes en displasia o hipoplasia renal, uni o bilateral, válvulas de uretra posterior, reflujo vesicoureteral, riñón poliquístico y estenosis a diferentes niveles.7
El retardo en el desarrollo psicomotor y la discapacidad intelectual no son comunes en estos pacientes. En algunos casos reportados con alteración en el desarrollo psicomotor, son atribuidos a un retardo en el diagnóstico de sordera. En otros afectados la discapacidad intelectual se ha asociado al hallazgo de alteraciones genéticas a nivel de la porción distal de 16q12 y 16q13, donde se encuentran, entre otros, el gen que codifica el transportador de noradrenalina (SLC6A2), capaz de interferir en el normal desarrollo cerebral del paciente.9
El diagnóstico diferencial se realiza de inmediato con la asociación VACTER (defectos vertebrales, anales, cardiovasculares, traqueoesofágicos, renales y de miembros, sobre todo radiales. Esto en ocasiones dificulta su identificación ya que la asociación malformativa tiene también expresividad variable y no permite distinguirlo el estudio minucioso de los defectos vertebrales.7,10
Se efectúa valoración de las malformaciones en el oído externo, en el pulgar y la región anorrectal con diagnóstico clínico.

