










 Serviços customizados
Serviços customizados Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
El síndrome Cockayne (CS), descrito por primera vez por un médico británico Edward Alfred Cockayne en 1933, las personas afectadas tienen un aspecto senil con sensibilidad a la luz solar y baja estatura. Su prevalencia estimada es de 1/500 000 nacidos vivos, con excepción de ciertas poblaciones aisladas o poblaciones endogámicas, donde se ha observado que aparece con una frecuencia mayor.1
Esta enfermedad tiene un patrón de herencia autosómico recesivo, por lo que la consanguinidad en los matrimonios aumenta el riesgo.2
Las características más sobresalientes en craneofaciales son; microcefalia, con cara pequeña, sin grasa, avejentada, ojos hundidos, nariz pequeña y afilada, dientes hipoplásicos, en ocasiones con ausencia de dientes permanentes, atrofia de los procesos alveolares y caries; piel: seca aspecto progeroide, pigmentada atrófica y con fotosensibilidad.
Síntomas neuropsíquicos: disfunción cerebelosa (ataxia, temblor, hipotonía), neuropatía periférica, discapacidad intelectual, postura encorvada con cifoescoliosis, limitación de la movilidad articular, frialdad y a veces cianosis en manos y pies con insuficiencia vascular periférica y arañas vasculares, la baja talla con disminución del tronco y aumento de la longitud de las extremidades. Síntomas menos destacados: criptorquidia, nefropatías túbulo-intersticial, hepatomegalia, hipertensión.3,4,5
Se reconocen cuatro tipos en dependencia del momento en que se manifieste; el CS Tipo I: los síntomas son progresivos y aparecen generalmente después de los 2 años de edad. Los afectados generalmente mueren antes de los 20 años; el CS Tipo II: comienza más temprano y puede estar presente desde el nacimiento. Los enfermos mueren antes de los siete años; el CS Tipo III: es una forma más tardía y menos severa que las anteriores. La esperanza de vida está en torno a los 40 o 50 años; el CS-XP, algunos casos de Síndrome Cockayne se manifiesta conjuntamente con la enfermedad Xeroderma pigmentoso; en esta condición incluye algunas características de ambas enfermedades: cáncer de piel, típico de los afectados por XP.5,6
Los afectados padecen problemas en el crecimiento y una degeneración multisistémica progresiva. La detención del crecimiento y el bajo peso es una de las manifestaciones más evidentes, ya que la mayoría de los pacientes no llegan a medir más de 1,15 m de altura ni a pesar más de 20 kg. El envejecimiento prematuro provoca anormalidades neurológicas causadas por la desmielinización difusa y progresiva del córtex cerebral.7,8
Por lo infrecuente de esta enfermedad y lo difícil de precisar cuándo comienzan los primeros signos clínicos, se decidió presentar este caso.
PRESENTACIÓN DE CASO
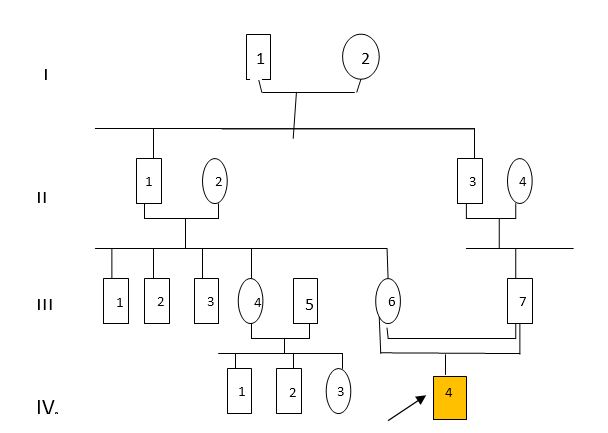
Paciente masculino de 14 años, desnutrido, con evaluación de mensuraciones: Talla/Edad 120cm, por debajo del tercer percentil para la talla; Peso/Edad 27kg por debajo del tercer percentil para el peso y la circunferencia cefálica 55cm se encuentra medidas normales. Descendiente de un matrimonio consanguíneo entre primos hermanos; esta familia reside en una localidad aislada de difícil acceso de la provincia Holguín con alta tasa de matrimonios consanguíneos. Se muestra el árbol genealógico de la familia, se muestran todos sus miembros; se aprecia solo un afectado, el descendiente de este matrimonio emparentado. (Fig. 1)
Atendido en consulta de endocrinología desde los cuatro años por fallo de medro con estudios hormonales normales. Fue valorado en consulta de dermatología por las lesiones atróficas causadas por fotosensibilidad, las cuales se presentaron como machas hiperpigmentadas e hipopigmentadas.
También evaluado por gastroenterología por abdomen globuloso, arañas vasculares con circulación colateral; hepatomegalia de 3cm sin cambios y estudios de función hepática normal.
Por último, se interconsultó con Genética Clínica; que durante la exploración descubrió despigmentación de pelo en estañas y cejas, el cabello resulta escaso y quebradizo. Además de la disminución del panículo adiposo, unido a una piel muy fina que permite observar la circulación venosa más acentuada en miembros inferiores causada por la insuficiencia venosa periférica, con manchas atróficas en todas sus regiones, esto incluye el abdomen, que al mismo tiempo se muestra voluminoso con numerosas arañas vasculares prueba de una circulación colateral y múltiples cicatrices por traumatismos y fotosensibilidad. (Fig. 2)
Para delinear el fenotipo es necesario un examen físico más minucioso y se le solicita a través de consentimiento informado a los padres los que aceden a que se examine nuevamente y tomarle fotografías para comparar con otros casos similares, pedir valoración de expertos en el tema y publicar en revistas científicas.
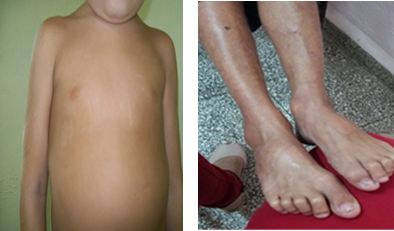
Fig. 2 Se aprecian el volumen del abdomen con las arañas vasculares de la misma forma en sus miembros inferiores.
La voz se percibió peculiar escandida, la conducta con hiperactividad que no le ha permitido integrarse al grupo escolar que le corresponde por su edad en su localidad.
Mantiene iguales características fenotípicas en el tiempo, se acentúa la insuficiencia vascular periférica, el ritmo de crecimiento muy disminuido; discapacidad intelectual ligera y retraso en el desarrollo puberal.
Se visualiza tronco y miembros superiores con disminución del panículo adiposo, una incipiente cifoescoliosis en columna torácica, además de escápulas aladas. También se puede apreciar la despigmentación del pelo, orejas grandes y el pobre panículo adiposo en la cara. (Fig. 3)
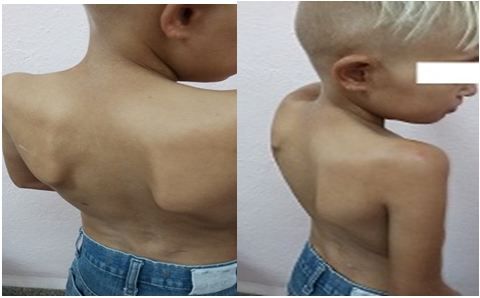
Sin embargo, el tamaño de las manos y los pies corresponde con su edad cronológica al igual que su edad ósea, con piel muy fina que deja visualizar la circulación capilar y venosa. (Fig. 4)

El retardo de la pubertad es otra distinción con genitales pequeños donde los testículos, el pene y la región púbica no se observan cambios puberales, con ausencia de vello púbica. Es apreciable la circulación venosa que resalta las características finas de la piel y la insuficiencia vascular. (Fig. 5)

Después de un exhaustivo interrogatorio y examen físico dismorfológico con la utilización del método clínico o de patrón se discuten todos los hallazgos en el colectivo de genética y con el equipo multidisciplinario que lo sigue en consulta, y se llega al diagnóstico clínico de que el paciente presenta un síndrome de envejecimiento prematuro, un Síndrome Cockayne tipo III.
DISCUSIÓN
El CS se manifiesta en distintos órganos y sistemas donde la piel que es un órgano expuesto muy dañado, el estudio de cultivo de fibroblastos puede mostrar la sensibilidad a la luz.1,2,3
Para el diagnóstico clínico en esta enfermedad genética se tienen en consideración los criterios mayores como son; falta de crecimiento posnatal (peso y talla en el percentil 5 o menos, a los dos años); disfunción neurológica progresiva que se manifiesta con retraso en el desarrollo temprano, seguido por deterioro del comportamiento e intelectual.
La resonancia magnética cerebral revela leucodistrofia y en algunos individuos se demuestran calcificaciones en la tomografía computarizada. Dentro de los criterios menores se describe fotosensibilidad cutánea y cabello friable, neuropatía desmielinizante periférica diagnosticada por electromiografía, test de conducción o biopsia de nervio, retinopatía pigmentaria (75 %) y/o cataratas (36 %), hipoacusia neurosensorial (60 %), caries dentales (86 %), apariencia física de “enanismo caquéctico” con adelgazamiento de la piel y el cabello, enoftalmos y una postura inclinada al pararse, hallazgos radiográficos característicos de engrosamiento del calvarium, epífisis escleróticas y anomalías vertebrales y pélvica. Ese paciente cumple con tres criterios mayores y cuatro menores.4,5
La degeneración del sistema nervioso central (SNC), puede manifestarse por desmielinización y discapacidad intelectual que puede progresar,6 como lo observado en este paciente con hiperactividad e inatención y se distrae con facilidad, además alteraciones en la memoria.
Las radiografías muestran engrosamiento del calvario craneal, posibles calcificaciones intracraneales, cifosis y escotadura anteriores de los cuerpos vertebrales dorsales.7,8 De la misma forma que se detalla en este paciente.
En esta enfermedad genética el tipo III, tiene una estrecha relación con el xeroderma pigmentoso (XP) ya que en múltiples ocasiones se presentan juntas. Debe realizarse diagnóstico diferencial con el síndrome Camat, el COFS y el Camfak (cataratas, microcefalia, fallo de medro, cifoescoliosis).7,8,9
Otros síntomas a destacar es la postura encorvada, con escoliosis y cifosis, la pérdida de la audición, la insuficiencia vascular periférica con temperatura corporal baja, orejas grandes.10,11 Este enfermo exhibe todas estas características fenotípicas.
Este síndrome puede ser provocado por mutación en dos genes, el ERCC6 (75 % de los casos) y el ERCC8 (CKN1) (25 % de los casos), localizados en los cromosomas 5q11 y 10q11 y que codifican para las proteínas CS-A y CS-B, respectivamente. 3,12
Estas proteínas están implicadas en la reparación del DNA, incluidas las lesiones provocadas por los rayos UV, con lo que una mutación en dichas proteínas provoca que las alteraciones producidas durante la transcripción no sean tan rápida y eficientemente reparadas como en células normales, con la consiguiente acumulación de DNA dañado. Esto es lo que provoca, entre otras cosas, el envejecimiento prematuro. También se ha asociado el síndrome a mutaciones de los genes XPB, XPD y XPG, en aquellos casos en que los pacientes presentan fenotipo combinado XP/CS. (8,11 )
Las mutaciones en el gen− ERCC8 (CKN1), son mutaciones patogénicas por cambios en el sitio de lectura, mutaciones sin sentido y pueden ser detectadas por análisis de las secuencias. El 30 % restante son deleciones grandes parciales del gen. Las mutaciones en el gen ERCC6, casi todas son mutaciones puntuales detectadas por secuenciación. La mayoría de estas son mutaciones que originan cambios en el sitio de lectura, que alteran la estructura primaria de la proteína y que provoca la pérdida de su función. 9,10,13
Este paciente se estudia desde los cuatro años por baja talla, se realizaron todos los estudios endocrinológicos, resultó estar en estándares normales y donde los exámenes dermatológicos coinciden con piel muy fina que permite observar la circulación capilar y que las biopsias informaron lesiones atróficas pigmentadas con acúmulo de melanina y en otras acrómicas por disminución de esta. También presenta disminución del panículo adiposo generalizado, que sugiere lipodistrofia a predominio de los miembros superiores e inferiores, que permite distinguir la circulación venosa con múltiples arañas vasculares; permitió llegar al diagnóstico de la insuficiencia venosa periférica que tiene el paciente; que unido a la discapacidad intelectual ligera y a los demás signo clínico anteriormente detallados, confirmó la sospecha clínica de un síndrome progeroide, específicamente el CS tipo III.
Aunque no está disponible en el país en los laboratorios de biología molecular el estudio de mutaciones para esta enfermedad, la impresión diagnóstica del equipo multidisciplinario determinó por el método clínico que esta es la enfermedad que padece este adolescente. Se decidió mantenerlo en consulta para mejorar su estado general con apoyo de otras especialidades que intervendrán en su seguimiento y tratamiento para elevar su calidad de vida. Se les brindó asesoramiento genético a los padres, a quienes se les explicó el riesgo de recurrencia en su descendencia.
CONCLUSIONES
Se considera de gran valor un examen físico exhaustivo que permita delinear correctamente el fenotipo; sin despreciar la importancia de un interrogatorio minucioso que facilite la elaboración del árbol genealógico que revele la relación de pertenezco de todos sus miembros. Durante el estudio de estas enfermedades de baja frecuencia con heterogeneidad clínica y genética; es necesario la intervención de un equipo multidisciplinaria para a través el método clínico lograr un acertado diagnóstico clínico.

