










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkCorSalud
versão On-line ISSN 2078-7170
CorSalud vol.9 no.4 Santa Clara oct.-dez. 2017
Unidad de Valoración del Riesgo de Muerte Súbita Familiar: Experiencia en la comunidad valenciana
Family Sudden Death Risk-Assessment Unit: Experience at the Valencian Community
Juan Giner Blasco1, Isabel Izquierdo Macián2, Esther Zorio Grima3
1. Instituto de Medicina Legal y Ciencias Forenses y Universidad Católica de Valencia. Valencia, España. Correo electrónico: giner_juabla@gva.es
2. Servicio de Pediatría. Hospital Universitario Politécnico La Fe. Valencia, España.
3. Servicio de Cardiología. Hospital Universitario Politécnico La Fe. Valencia, España.
RESUMEN
La muerte súbita se define como el fallecimiento inesperado que acontece antes de una hora desde el inicio de los síntomas, este tipo de muerte tiene un alto impacto social, mediático y económico. La primera causa es la de origen cardíaco y dentro de estas la cardiopatía isquémica es la más frecuente, pero las cardiopatías familiares (canalopatías y miocardiopatías) son porcentualmente más importantes en niños y jóvenes, donde representan la primera causa de muerte súbita cardíaca. Estas cardiopatías familiares tienen un claro sustrato genético que justifica la indicación de un adecuado estudio de los familiares de los fallecidos. De acuerdo a los datos de la población española del censo de 2013 (46,7 millones de habitantes) en la comunidad valenciana, que representa el 10% de esta población, se estima que residen 20000 personas con alguna cardiopatía familiar potencialmente letal. Dada la importancia y el impacto social de la muerte súbita de origen cardíaco, y puesto que la autopsia médico-legal tiene limitaciones para diagnosticar la enfermedad subyacente en este tipo de muertes, la estrategia más oportuna es el enfoque multidisciplinar, motivo por el cual en el año 2008 se creó la Unidad de Muerte Súbita Familiar y Cardiopatías Familiares en esta región.
Palabras clave: muerte súbita cardíaca, cardiopatías heredadas, factores de riesgo, diagnóstico, prevención de enfermedades.
ABSTRACT
The sudden death is defined as the unexpected death that occurs within an hour of the onset of symptoms. This type of death has a high social, media and economic impact. The first cause is of cardiac origin, and within this, the ischemic heart disease is the most frequent, but family heart diseases (channelopathies and cardiomyopathies) are more important in children and young people, where they represent the first cause of sudden cardiac death. These family heart diseases have a clear genetic substrate that justifies the indication of an adequate study of the relatives of the deceased. According to the data of the Spanish population of the 2013 census (46.7 million inhabitants) in the Valencian Community, which represents 10% of this population, it is estimated that there are 20.000 people with some potentially lethal heart disease. Given the importance and the social impact of sudden death of cardiac origin, and since the medical-legal autopsy has limitations to diagnose the underlying disease in these types of deaths, the most opportune strategy is the multidisciplinary approach, which is why in 2008, the Family Sudden Death and Family Heart Diseases Unit was created in this region.
Key words: sudden cardiac death, inherited heart disease, risk factors, diagnosis, disease prevention.
INTRODUCCIÓN
La muerte súbita (MS) se define como el fallecimiento inesperado que acontece antes de una hora desde el inicio de los síntomas, este tipo de muertes tiene un alto impacto social, mediático y económico1.
La primera causa de MS es la de origen cardíaco y dentro de estas la cardiopatía isquémica es la más frecuente. Sin embargo, las cardiopatías familiares (canalopatías y miocardiopatías) son porcentualmente más importantes en niños y jóvenes, donde representan la primera causa de MS cardíaca2 (Figura 1). A diferencia de la cardiopatía isquémica, las cardiopatías familiares tienen un claro sustrato genético que justifica la indicación de un adecuado estudio de los familiares de los fallecidos con ampliación del círculo a estudio en cascada, según los resultados en las generaciones previas.
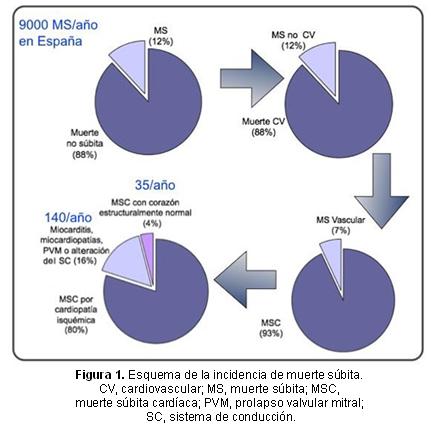
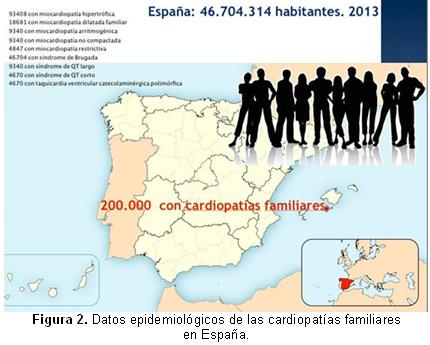
Todas las cardiopatías familiares, excepto la miocardiopatía hipertrófica que tiene una prevalencia de 1/500, tienen prevalencias menores a 1/2000-5000, que encajan en la definición de enfermedad rara. Atendiendo a la población española del censo de 2013 (46704314 habitantes) y a las prevalencias de estas enfermedades, en nuestro país se estima que hay unos 200000 sujetos con cardiopatías familiares (Figura 2), hayan sido diagnosticadas y tratadas (una minoría) o no (desconocedores de la enfermedad y desprotegidos frente a eventos adversos)3. Si la población de la Comunidad Valenciana representa el 10% de la población española, en esta comunidad autónoma se estima que residen 20000 habitantes con alguna cardiopatía familiar potencialmente letal.
Dada la importancia e impacto social de la MS de origen cardíaco y puesto que la autopsia médico-legal, en este tipo de muertes, tiene limitaciones para diagnosticar la enfermedad subyacente, la estrategia más oportuna parece ser un enfoque multidisciplinar, motivo por el cual en el año 2008 se creó la Unidad de Muerte Súbita Familiar y Cardiopatías Familiares en la comunidad valenciana.
CARACTERÍSTICAS FUNCIONALES
Esta unidad es pionera en España en el abordaje multidisciplinar y multiinstitucional de las familias afectadas con MS cardíaca, especialmente aquellas relacionadas con cardiopatías familiares de origen genético. De hecho, es la primera unidad multidisciplinar en España con acuerdo explícito entre las Consellerías de Sanidad (de quien dependen los clínicos e investigadores) y Justicia (de quien dependen los médicos forenses y los patólogos forenses), de forma similar a como previamente se ideó en el Reino Unido (Chapter Eight, NHS 2005)4. La Unidad cuenta con una subunidad forense y otra hospitalaria (Figura 3).
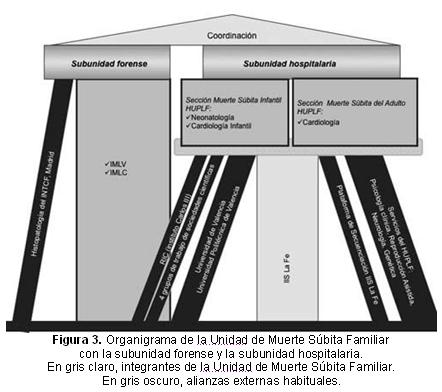
Grupos de trabajo de sociedades científicas: «Cardiopatías Familiares» de la Sociedad Española de Cardiología, «Cardiomyopathies » de la Sociedad Europea de Cardiología, «Muerte Súbita Infantil» de la Asociación Española de Pediatría y «Muerte Súbita» de la Sociedad Española de Patología Forense.
HUPLF, Hospital Universitario y Politécnico La Fe; IMLC, Instituto de Medicina Legal de Castellón; IMLV, Instituto de Medicina Legal de Valencia; INTCF, Instituto Nacional de Toxicología y Ciencias Forenses; ISS, Instituto de Investigación Sanitaria; RIC, Red de Investigación Cardiovascular.
En la subunidad forense se realizan la autopsias judiciales que tienen como fin, entre otros, identificar la causa de la muerte, proporcionar datos morfológicos y la recogida de muestras biológicas. Entre 2008 y 2015, se han estudiado 618 casos de MS cardíaca. Según los resultados de las autopsias las MS de este tipo se clasifican en los siguientes grupos:
1. Corazón estructuralmente normal (donde hay que sospechar canalopatías).
2. Miocardiopatías (hipertrófica, dilatada, no compactada, arritmogénica o no determinada).
3. Disección de aorta torácica no ateromatosa (incluye aorta bicúspide).
Las familias de los fallecidos de estos grupos son citadas en los Institutos de Medicina Legal donde se les informa el resultado de la autopsia y se les ofrece la posibilidad del estudio familiar hospitalario (subunidad clínica). El protocolo de actuación se detalla en la figura 4.

Las muertes súbitas isquémicas se estudian igualmente en las autopsias que se realizan en los Institutos de Medicina Legal. Sin embargo, dada la base poligénica y multifactorial de la aterosclerosis, los familiares de los fallecidos no son valorados en la subunidad clínica, sino que se les recomienda que controlen estrictamente sus factores de riesgo cardiovascular y, en caso de sintomatología, sean remitidos a su cardiólogo.
Se han establecido circuitos para el flujo de las muestras biológicas procedentes de la autopsia que van a ser estudiadas dentro del ámbito asistencial, investigativo, o ambos; en particular, las de plasma post mortem para estudio de perfil lipídico y las que se utilizan para estudios genéticos en el contexto de las cardiopatías familiares de causa genética.
Una vez que el familiar firma el consentimiento son remitidas a la subunidad clínica donde se realiza un estudio que incluye pruebas clínicas y genéticas.
PROTOCOLO DE ACTUACIÓN
El protocolo de esta unidad, nutrido _en gran medida_ de la experiencia previa de la literatura, incluye estudios para los familiares de primer grado que se extienden en cascada ampliando el árbol familiar, según los resultados1-8.
Los estudios clínicos en los familiares dependen de la clasificación del individuo en estudio (probando) y, debido al gran desarrollo de la cardiogenética en los últimos años _donde se han publicado guías prácticas para la realización de exámenes genéticos en el contexto de estas enfermedades1-3_, se complementan con los resultados de los estudios genéticos. Dichos estudios se realizan en ADN extraído de la sangre del fallecido (obtenida en la autopsia). Si no se dispone de esta, se elige a un familiar con el mismo fenotipo que aquel y, solo en caso de no haberlo, no se realizarían los estudios genéticos.
Hasta ahora, algo más del 25% de los integrantes de familias con cardiopatías familiares han recibido un diagnóstico (clínico, genético, o ambos) que ha conllevado la modificación de hábitos de vida (cambios en actividad deportiva), iniciar el uso de fármacos (fundamentalmente betabloqueantes), proporcionar un listado de medicamentos a evitar (por ser potenciales desencadenantes de arritmias en su enfermedad), mantener revisiones periódicas o, incluso, implantar un desfibrilador automático (71 dispositivos implantados en estas familias).
Los diagnósticos más frecuentes han sido miocardiopatía arritmogénica, hipertrófica, dilatada y no compactada; cardiopatía por laminopatía, síndromes de QT largo y de Brugada, y taquicardia ventricular catecolaminérgica polimórfica.
El reconocimiento de estas cardiopatías familiares ha permitido ofrecer consejo clínico y genético a las parejas con deseo gestacional, en dependencia de la información disponible en cada caso.
CONFLICTOS DE INTERESES
Los autores declaran que no existen conflictos de intereses.
REFERENCIAS BIBLIOGRÁFICAS
1. Gollob MH, Blier L, Brugada R, Champagne J, Chauhan V, Connors S, et al. Recommendations for the use of genetic testing in the clinical evaluation of inherited cardiac arrhythmias associated with sudden cardiac death: Canadian Cardiovascular Society/Canadian Heart Rhythm Society joint position paper. Can J Cardiol. 2011;27:232-45.
2. Charron P, Arad M, Arbustini E, Basso C, Bilinska Z, Elliott P, et al. Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2010;31:2715-26.
3. Barriales-Villa R, Gimeno-Blanes JR, Zorio-Grima E, Ripoll-Vera T, Evangelista-Masip A, Moya-Mitjans A, et al. Protocolo de actuación en las cardiopatías familiares: síntesis de recomendaciones y algoritmos de actuación. Rev Esp Cardiol. 2016;69:300-9.
4. Coronary Heart Disease Team. National Service Framework for Coronary Heart Disease - Chapter Eight: Arrhythmias and Sudden Cardiac Death. London: National Health Service; 2005.
5. Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 2011;8:1308-39.
6. Zipes DP, Camm AJ, Borggrefe M, Buxton AE, Chaitman B, Fromer M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death). Developed in Collaboration With the European Heart Rhythm Association and the Heart Rhythm Society. J Am Coll Cardiol. 2006;48:e247-346.
7. Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35:2733-79.
8. Bhonsale A, James CA, Tichnell C, Murray B, Gagarin D, Philips B, et al. Incidence and predictors of implantable cardioverter-defibrillator therapy in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy undergoing implantable cardioverter-defibrillator implantation for primary prevention. J Am Coll Cardiol. 2011;58:1485-96.
9. van Rijsingen IA, Arbustini E, Elliott PM, Mogensen J, Hermans-van Ast JF, van der Kooi AJ, et al. Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a European cohort study. J Am Coll Cardiol. 2012;59:493-500.
Recibido: 18 de abril de 2017
Aceptado: 18 de mayo de 2017
Juan Giner Blasco. Instituto de Medicina Legal y Ciencias Forenses y Universidad Católica de Valencia. Valencia, España. Correo electrónico: giner_juabla@gva.es

