










 Serviços customizados
Serviços customizados Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La presente investigación se origina dentro del proyecto Estudio Computacional de Virus en Microfluidos, que forma parte de la colaboración entre la Universidad de las Ciencias Informáticas (UCI), el Centro de Ingeniería Genética y Biotecnología (CIGB) y el grupo SciVis del Consejo Nacional de Investigaciones (Consiglio Nazionale delle Ricerche) de Milán, Italia. Como parte de este proyecto, se investigan propiedades dinámicas en la sangre y otros microfluidos de virus como el dengue y el zika, con el fin de obtener información sobre el proceso de infección de estos patógenos en el organismo, frecuentemente asociados a casos graves en las enfermedades que producen. Debido al alto impacto en la salud pública de estos virus, todo nuevo conocimiento que se obtenga puede resultar de gran relevancia social. Adicionalmente, estos nuevos conocimientos pueden derivar en el desarrollo de fármacos, que no solo impactarían en la salud de las personas, sino también en la economía, al convertirse en rublos exportables.
En cuanto a la investigación, entre las líneas a desarrollar dentro del proyecto, se incluye la visualización científica de los resultados de las simulaciones obtenidas por dinámica molecular, de los sistemas moleculares que comprenden los virus y las moléculas del plasma sanguíneo. Primeramente, cuando se habla de simulación, esta se refiere al proceso de diseñar un modelo de un sistema real, imponer un conjunto de criterios para las operaciones y, realizar experimentos, con el fin de comprender el comportamiento del sistema o evaluar nuevas estrategias -dentro de los límites impuestos- (Shannon and Johannes, 1976).
La información recopilada sobre las propiedades dinámicas de las macromoléculas o de los sistemas moleculares (formados por varias moléculas interactuantes) es lo suficientemente rica como para cambiar el paradigma habitual de la bioinformática estructural, de estudiar estructuras individuales para analizar con- juntos conformacionales (Hospital et al., 2015). Una limitación importante en el procedimiento general de corrida/análisis de la dinámica molecular es la imposibilidad de ejecutar una simulación y visualizar sus re- sultados simultáneamente (en tiempo real), debido al costo computacional de ambos procesos. Situación que obliga a configurar las simulaciones, ejecutarlas y, posteriormente visualizar y analizar el resultado. Estos procesos se pueden llevar a cabo en varias iteraciones, hasta que se obtengan resultados que aporten nuevos conocimientos. Además, el análisis de los resultados obtenidos por esta vía se suele realizar a partir de abstracciones complejas, que dificultan su interpretación, al alejarse de representaciones que resulten intuitivas para el investigador.
La visualización científica, por su parte, es una rama interdisciplinaria de la ciencia que consiste en la transformación de datos abstractos en imágenes (Friendly et al., 2001). Se hace con los fines de evitar que da- tos importantes sean omitidos por equivocación, facilitar la comprensión de conceptos complicados, indicar qué datos son importantes y cuáles no lo son, así como simplificar la comunicación entre los científicos. La razón principal para visualizar, es la tesis de que el sentido más desarrollado de los humanos es la vista y por eso es la manera más fácil de comunicar la información, especialmente cuando es compleja o viene en grandes cantidades. Este es el caso de la visualización de simulaciones de dinámica molecular, que forma parte de un área más general que es la visualización molecular, que implica representar modelos moleculares parar poder explorarlos y comprenderlos.
Con el avance de las tecnologías gráficas por computadora, se crearon herramientas para la visualización de estructuras moleculares en tres dimensiones, por ejemplo, VMD (Humphrey et al., 1996), SPDBViewer (Guex and Peitsch, 1997), Chimera (Pettersen et al., 2004), PyMOL (DeLano, 2002) y demás, las cuales permiten la lectura de la información proveniente del repositorio público Protein Data Bank (PDB) (Bernstein et al., 1977). A partir de los archivos de moléculas, que contienen las coordenadas tridimensionales de cada átmo de ellas, estos programas tienen la capacidad de leerlos y lograr la representación de las moléculas en el espacio 3D, ofreciendo la posibilidad de interactuar con los modelos moleculares, rotando, trasladando y modificando el grado de amplificación de la imagen, así como los formatos de visualización, entre otras funciones. De estos, PyMOL y VMD ofrecen recursos para visualizar trayectorias provenientes de simulaciones moleculares, pero requieren del usuario cierta experiencia en el manejo de los programas. En el caso de VMD, se encuentran interconectados a él, programas dedicados específicamente para la construcción y análisis de superficies moleculares de sistemas nanoscópicos, como es el caso de NanoShaper (Decherchi et al., 2018). El campo de la animación científica se puede explotar para facilitar el estudio de fenómenos complejos, combinando las herramientas de animación e infografía, con información científica, e incluyendo conceptos asociados al arte, que faciliten su comprensión. Entre las herramientas más utilizadas, se encuentra el arte y la ciencia de la animación 3D, la cual consiste en la creación y animación de objetos en el espacio tridimensional (con superficies, esqueletos y propiedades físicas simuladas) en un mundo virtual, que se pueden “filmar” utilizando cámaras y luces virtuales (Andrei et al., 2010). Hay varios programas disponibles para esto, incluidos los paquetes comerciales Maya / Autodesk, 3D Studio Max y Softimage XSI (todos de Autodesk) (Auto desk, 2021) y el software de modelado 3D de código abierto Blender (Blender Foundation, 2021). Este último es uno de los motores más potentes de Computer Graphics (CG) para la gestión de contenido 3D, es decir, creación, animación, texturizado y, renderizado de objetos y escenas visuales. Blender es un potente entorno de desarrollo dedicado a la creación de imágenes y animaciones 3D, cuya principal ventaja respecto a otras herramientas similares como las antes mencionadas, es que se trata de una aplicación totalmente gratuita, la cual al ser multiplataforma y de código abierto, permite a los usuarios introducir nuevas características, como, por ejemplo, la introducción de nuevos Add-Ons.
En base a este último fue desarrollado BioBlender, un programa de código abierto, de distribución gratuita, multiplataforma, interoperable y compatible de animación 3D, efectos visuales y videojuegos. Donde la combinación de infografías 3D, motor de juegos y programas científicos, hacen de BioBlender un completo instrumento para elaborar el movimiento de proteínas y mostrar sus características superficiales mediante un código visual, basado en texturas y efectos especiales de partículas. De esta forma es posible mostrar las pro- piedades físicas y químicas de una molécula tanto en movimiento como estática (Andrei et al., 2010, 2012; Zini et al., 2012). Permite la representación intuitiva de propiedades de superficie de las biomoléculas, mos- trando su superficie de manera fotorrealista, permitiendo así la visualización de complejas propiedades de superficies de proteínas como el potencial electrostático (EP) y potencial lipofílico molecular (MLP). Otro aporte de BioBlender es la posibilidad de representar sistemas moleculares complejos, en un entorno que reproduzca el interior de una célula, mediante la combinación de simulaciones de varias moléculas y componentes celulares de membrana. Diversas investigaciones demuestran el potencial de este módulo, ‘Intuitive representation of surface properties of biomolecules using BioBlender’ (Andrei et al., 2012) y ‘The Represen- tation of Electrostatics for Biological Molecules’ (Zoppe` and Loni, 2015), son ejemplo de ellas.
Una de las principales desventajas de BioBlender en su estado actual, es la dificultad del uso de las herra- mientas que posee, causado por los constantes cambios que se realizan en la plataforma en la que está basado. BioBlender fue desarrollado por el grupo SciVis para una versión ya caducada de la suite de creación 3D Blender, y no se ha seguido actualizando hasta la fecha, por lo que su actualización es uno de los propósitos del proyecto dentro del cual se basa la presente investigación. El objetivo es desarrollar una nueva versión de BioBlender funcional en versiones actuales de Blender, lo que conlleva a la actualización y reestructuración de su código y, un estudio exhaustivo de la documentación de la plataforma para conocer los nuevos cambios que presenta. Vinculado a esto y sin posibilidad de separación, se encuentra la migración de su código a versiones vigentes del lenguaje de programación Python, lenguaje en el que está basado tanto Blender como BioBlender.
MÉTODOS O METODOLOGÍA COMPUTACIONAL
Para el presente trabajo se tuvieron en cuenta modelos extraídos de la base de datos biológica Protein Data Bank, los cuales están contenidos en archivos con formato PDB, conformados por un conjunto de átomos con identidad y posición, unidos entre sí por enlaces químicos. BioBlender tiene la capacidad de leer dichos archivos y obtener la información necesaria para representar biomoléculas en el espacio 3D. Entre los modelos utilizados con mayor frecuencia para la realización de las pruebas en los cambios realizados en el código, se encuentran los siguientes:
6R2X (Estructura obtenida por Resonancia Magnética Nuclear (RMN) de Cromogranina A (F39-D63). Pequeño péptido compuesto por 25 aminoácidos, que comprenden 214 átomos (436 incluido los hidrógenos))
1BNA (Estructura de un dodecamer de ADN-B. Conformación y dinámica. Compuesta de 566 átomos que conforman 12 pares de bases)
1CFC (Calmodulina (CaM) sin calcio. CaM es una proteína bien estudiada compuesta por 148 aminoácidos, que comprende 1166 átomos (2262 incluidos los hidrógenos), bien conservada a lo largo de la escala evolutiva)
6LU7 (La estructura cristalina de la proteasa principal COVID-19 en complejo con un inhibidor N3. Compuesta por 2500 átomos, los cuales conforman 312 aminoácidos).
4IHV (Estructura cristalina de Fis, unida a la secuencia de ADN F28 de 27 pb (AAATTTGTTT- GAGCGTTGAGCAAATTT). Editado para eliminar proteínas y dejar solo el ADN, por SciVis, para el estudio de ADN con BioBlender)
1L9K (Dengue Metiltransferasa. Contiene 2158 átomos, los cuales conforman 261 aminoácidos)
3IV5 (Estructura cristalina de Fis unida a la secuencia de unión óptima F1 de 27 pb. Contiene 2618 átomos, los cuales conforman 243 aminoácidos)
1A4U (Alcohol Deshidrogenasa de Drosophila Lebanonensis. Compuesto por 508 aminoácidos, con un total de 4091 átomos)
1ATN (Estructura atómica de la Actina: complejo DNASE I. Es una proteína con mayor extensión, está compuesta por 5019 átomos, los cuales conforman 631 aminoácidos)
Para lograr la migración del código de Python v2.6 a Python v3.7 en adelante, se hizo uso del Entorno de Desarrollo Integrado (IDE) PyCharm Community v2020.2.5, disponible en https://www.jetbrains.com/es-es/pycharm/download/, el cual proporciona una amplia gama de herramientas. Con la in- corporación al mismo como intérprete a la versión 3.7.5 de Python, se ha llevado a cabo el objetivo de la
investigación, donde se analizó el código fuente, para luego dividirlo en diferentes módulos, siguiendo las
normas básicas de Python PEPs, las nuevas propuestas de mejora de Python para cada versión y los nuevos cambios en propiedades y métodos vinculados con el lanzamiento de Blender v2.80, expuestos en la documentación de la API de Python de Blender v2.80 (Blender Foundation, 2019).
La incorporación de un Sistema de Control de Versiones (VCS) a PyCharm, permite a los desarrolladores llevar un control estricto de sus versiones y poder volver con total seguridad a cada una de ellas cuando se presenten problemas posteriores en el desarrollo del sistema (Wanumen and Silva, 2008). Se ha utilizado esta herramienta para el control de versiones de BioBlender, lo cual ha facilitado la modificación y actualización del código, ya que ha permitido tener un historial de cambios o versiones para su reutilización en caso de que se necesiten. PyCharm no solo cuenta con un sistema de control de versiones lo- cal, sino que permite la vinculación con GitHub, el servicio basado en la nube que se encuentra aloja- do en https://github.com e implementa a Git, un VCS distribuido de código abierto y gratuito, diseñado para manejar todo, desde proyectos pequeños a muy grandes con velocidad y eficiencia (Git, 2021). La utilización de un VCS como GitHub, ha permitido la colaboración de desarrolladores de diferentes lu- gares del mundo y, además, ha posibilitado que investigadores hagan pruebas con BioBlender y mencio- nen sus inquietudes o errores obtenidos, facilitando el trabajo de corrección de errores en las nuevas ac- tualizaciones. Los códigos del desarrollo, manual de instalación y guía de usuario, pueden encontrarse en https://github.com/PabloEnmanuelRamos/BioBlender21.git.
Métodos de pruebas de software
El proceso de pruebas de software constituye uno de los aspectos fundamentales para medir el estado de calidad de un sistema informático. En el caso de BioBlender, para la realización de las pruebas de software se aplicaron pruebas funcionales, mediante métodos de Caja Blanca para la evaluación de las funcionalidades internas, y Caja Negra para la evaluación de las funcionalidades externas. Como apoyo a las pruebas funcionales, a través del método de Caja Negra, se aplicaron pruebas de Usabilidad mediante el método de Inspección. Para la ejecución de estas pruebas en la presente investigación se realizó un análisis de las principales bibliografías especializadas en el tema, profundizando en las diferentes técnicas de pruebas que existen, fundamentalmente en las técnicas de los métodos utilizados en la presente investigación.
Se utilizó la técnica de partición de equivalencia del método de Caja Negra para dividir los datos de entrada en diferentes tipos, para así determinar diversos casos de pruebas y comprobar funciones específicas con cada uno de ellos. Los casos de prueba se agruparon en dependencia de la estructura del modelo ya que para lograr su correcta representación en el espacio 3D se tienen que pasar por ciertos algoritmos y por medio de ellos ir creando su estructura, así como sus enlaces entre cada átomo y grupos de átomos. Sumado a esto se utiliza la técnica Heurística del método de Inspección para evaluar la usabilidad y así comprobar principios de diseño usable (principios heurísticos) previamente establecidos. Estos principios son directrices que establecen requisitos que debe cumplir el diseño con el fin de facilitar su comprensión y uso por el usuario final. Para el caso de la presente investigación se utilizó como herramienta de apoyo a este tipo de prueba, la lista de chequeo establecida por el Departamento de Calidad de la UCI, definida como un listado de preguntas, en forma de cuestionario, utilizado para verificar el grado de cumplimiento de determinadas reglas heurísticas establecidas a priori con un fin determinado.
Mediante el método de Caja Blanca se investigó en la estructura interna del código, realizando un seguimiento del código fuente según se van ejecutando los casos de pruebas, determinándose de manera concreta las ins- trucciones que han sido ejecutadas por estos. Para ello se desarrollaron casos de pruebas específicos buscando la ejecución de cada ruta posible del módulo. Se utilizaron pruebas de cobertura lógica como de cobertura de caminos y de cobertura de Condición/Decisión, además, de estructura de datos locales. Por medio de estos tipos de prueba se hizo un estudio de todas las variables del módulo, evitando la declaración de variables con el mismo nombre, declaradas local o globalmente, analizando su comportamiento. Mediante la cobertura de caminos se destinaron suficientes casos de pruebas para ejecutar todas las instrucciones de BioBlender, comprobando cada una de sus funciones y obteniendo de estas los errores producidos durante las ejecuciones, para luego determinar su posible solución.
Método para la visualización del Potencial Lipofílico de la Molécula (MLP)
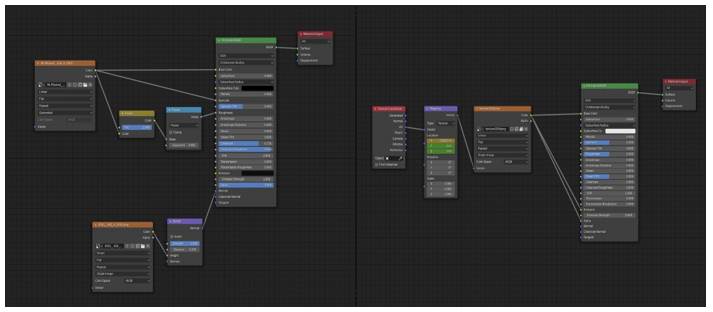
Debido a los cambios estructurales ocurridos a partir de la versión 2.8 de Blender, la representación del cálculo del MLP fue imposible de realizar, por lo que hubo que esperar a la salida de Blender 2.92, el cual trajo consigo la posibilidad de hornear la textura con colores de vértices. Se hizo una reestructuración del código del módulo de BioBlender que corresponde a esta función, actualizándolo a las condiciones actuales, con el objetivo de lograr el horneado de la textura que antes era imposible. Posteriormente se desarrolló un material para la superficie de la molécula con la utilización de la herramienta Editor de Sombreado, brindada por Blender para la creación de materiales utilizando los llamados nodos de sombreado, los cuales generan valores, vectores, colores y sombras. Mediante el uso del nodo Image Texture se jugó con la imagen del resultado de hornear la textura de la superficie con color de vértices, proceso que necesita un Mapeo UV de la superficie generada de la molécula, proyectar una malla tridimensional (x,y,z) en una imagen bidimensional (x,y). Además, se utilizó el resultado de la composición de la imagen con una textura predeterminada que representa la rugosidad de la molécula, ubicada en la carpeta de datos de BioBlender. El material generado, fue guardado en un archivo con formato BLEND, funcionando como una biblioteca de donde se puede extraer las propiedades dadas a un objeto en específico, así como extraer el objeto, sus materiales y demás. Creada con el propósito de obtener el material para luego utilizarlo en cada superficie que el usuario genera con el objetivo de representar el potencial lipofílico de la molécula en su superficie. En la Figura 1 se puede observar del lado izquierdo el enlace entre los nodos generados para lograr una superficie fotorrealista.
Método para la representación del Potencial Electrostático (EP)
Otro de los problemas que trajo consigo las nuevas versiones de Blender es la no representación de sistemas de partículas provenientes de un emisor al renderizar los objetos de la escena. Por ende, la forma de representación de las partículas varió, ya que las curvas por las que las partículas se desplazaban se han convertido a objeto, y se les ha proporcionado un material, creado en el anteriormente mencionado Editor de Sombreado. Se diseñó una textura de imagen en blanco y negro, de forma intercalada por columnas para asignárselo a la curva. Además, se utilizó el nodo Texture Coordinate para obtener las coordenadas de la textura del Mapeo UV realizado a la curva, para ser utilizado como vector de entrada del nodo Mapping, en donde el valor de la localización en el eje X varía debido a la aplicación de una animación a dicha propiedad, con la inserción de un fotograma clave, lo que provoca que su valor cambie a medida que pasa el tiempo, ocurriendo una interpolación entre dos de sus propiedades animadas, posibilitando que la textura creada se desplace por la curva y simule partículas trasladándose sobre ellas (de positivo a negativo). A esta curva de tiempo se le denomina Curvas-F, a la cual se le agregó, además, el modificador Cycles para lograr un ciclo repetitivo de su traslación. El material generado fue guardado en una biblioteca de Blender, para obtenerlo y asignárselo a cada curva que se genera en el cálculo del EP. En la Figura 1 se puede observar del lado derecho el enlace entre los nodos que posibilitan la representación de las partículas.
RESULTADOS Y DISCUSIÓN
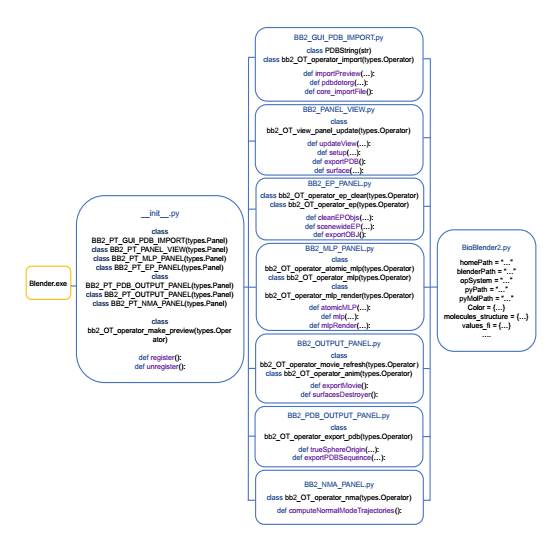
El primer paso para el desarrollo de una nueva versión de BioBlender funcional en las nuevas versiones de Blender, es la migración de su código a una versión vigente de Python, obteniéndose a la vez, una reestructu- ración del script que acoplaba todas las clases y funciones. Se hizo modular su código, dividiendo en varios módulos el script original. Cada uno de ellos contiene una función específica a desarrollar, proporcionando un mayor control y un mejor acceso a las clases y funciones. La Figura 2 muestra una perspectiva general del código fuente, donde se puede apreciar cada uno de los módulos resultantes.
Teniendo en cuenta lo anterior, se llevó a cabo la tarea de hacer que cada uno de los módulos que conforman BioBlender sean registrados satisfactoriamente en Blender y, una vez logrado, se continuó a la importación de un modelo y a obtener de este, sus distintas representaciones en el espacio tridimensional. BioBlender da la posibilidad de visualizar solo lo que el usuario desee ver, de una forma sencilla y rápida, sin la necesidad de conocimientos previos de la plataforma. Es posible importar una variedad de estructuras sin recibir fallos en la importación, obteniendo, además, todas sus conformaciones, las cuales posibilitan crear una secuencia relativa de su movimiento, sin tener en cuenta sus propiedades físico-químicas. En la Figura 3 se muestra un ejemplo de las distintas formas de visualizar una proteína en el espacio 3D, según las opciones que brinda BioBlender.

Fig. 3 Proteína CaM en el espacio 3D. Cadena principal → + Cadenas laterales → + Hidrógenos (solo en caso ser importados) → Superficie de la proteína.
Para la visualización de las propiedades de superficie de las biomoléculas, las dificultades estaban relaciona- das con la nueva forma de texturizado de Blender, basado en nodos. El objetivo es representar el potencial lipofílico atribuyendo los valores a cada vértice de la superficie. El potencial depende de las propiedades de los átomos inmediatamente debajo de la superficie. Estos se integran mediante varios pasos (Andrei et al., 2010), y finalmente se renderizan, utilizando el método expuesto en la sección anterior, que atribuye el valor a los vértices de la malla de la superficie generada y permite una visualización fotorrealista detallada del MLP. Los resultados obtenidos se muestran en las Figura 4 y Figura 5.

Fig. 4 Relieve (por defecto) Textura calculada con el horneado a color de vértices de la proteína CaM Textura final de la proteína CaM calculada de la composición de las imágenes anteriores.
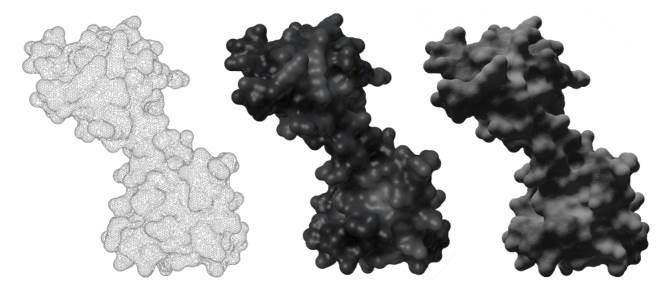
Fig. 5 Proteína CaM. Malla → Visualización de MLP como niveles de gris (el blanco es muy hidrofóbico, el negro es muy hidrofílico) → Visualización de MLP.
Una serie similar de obstáculos se encontró durante la actualización de la función “Visualización EP” (EP Visualization), la cual simula partículas de líneas que fluyen a lo largo de las líneas de campo (de positivo a negativo) proporcionalmente a la carga total de la proteína. La representación del EP como partículas en movimiento en una trayectoria, reproducidas en el tiempo, se interpreta fácilmente y transmite la idea de polaridad de las áreas cargadas de una biomolécula. En imágenes fijas, las partículas lineales no pueden transmitir la polaridad de las áreas cargadas de las moléculas, y debe sustituirse por formas que transmitan información sobre la dirección, como pequeños cometas o flechas. En la Figura 6, se puede observar el ejemplo del Alcohol Deshidrogenasa de Drosophila Lebanonensis (PDB 1A4U) en el lado izquierdo y la Actina (PDB 1ATN) en el extremo derecho, como un solo fotograma de una representación animada.
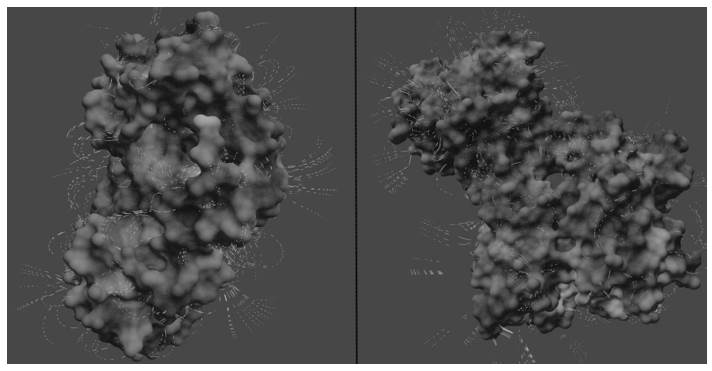
Fig. 6 Representación de un solo fotograma de la animación. Lo que impide al ser una imagen fija, que las partículas transmitan la polaridad de las áreas cargadas de las moléculas.
Representar de forma visual las características de la superficie de las proteínas en movimiento, permite una percepción inmediata del comportamiento de sus características moleculares. Por lo que otra de las funciones actualizadas correctamente es “Exportar Película” (Movie Output). Básicamente, esta permite obtener una secuencia relativa del movimiento de las proteínas en la escena, ya que por cada fotograma se exporta una imagen renderizada, de forma tal que al unir esta secuencia de imágenes se crea una película. Las moléculas se pueden renderizar acorde a las distintas formas de representación anteriormente expuestas y, además, representando el MLP en su superficie, en conjunto con el EP. Cuando se genera una película en donde se calculan el EP y el MLP, se necesita tiempo, ya que para lograr su representación, es indispensable la ejecución de varios programas que generan archivos necesarios, además de los cálculos de las líneas de campo, creación de las imágenes de texturas y su asignación a cada malla. A continuación, en la Figura 7 se visualizan algunos de los modelos de la Cromogranina A (PDB 6R2X).

El Análisis de Modos Normales (NMA) puede facilitar la investigación rápida y sistemática de la dinámica de las proteínas utilizando datos provenientes de la base de datos biológica Protein Data Bank, ya que representa una descripción de las vibraciones de una proteína en términos de osciladores armónicos. BioBlender cuenta con la posibilidad de calcular la trayectoria del NMA, para ello, es necesaria la utilización de la librería Prody, la cual permite realizar dicho análisis. Esta función fue actualizada con éxito y, una vez calculada la trayectoria, se exporta un archivo en formato PDB, listo para ser importado y reproducir su movimiento.
Para evaluar la calidad de la lógica interna y externa de BioBlender, se llevaron a cabo 2 iteraciones de pruebas de software, hasta alcanzar resultados satisfactorios, atendiendo al correcto comportamiento del sistema ante diferentes situaciones. A modo resumen:
Pruebas funcionales aplicando técnica de pruebas de Caja Negra: se obtuvo un total de 9 defectos en la primera iteración, 6 clasificados en el grupo de Opciones que no funcionan, 2 de Validación y 1 de Ortografía.
Pruebas de usabilidad utilizando la lista de chequeo para aplicar la técnica Heurística: se obtuvo en la primera iteración un total de 10 defectos, 5 de estos pertenecen a Visibilidad del sistema, 4 a Estética y diseño minimalista y, 1 a Consistencia y estándares.
Pruebas funcionales aplicando técnica de pruebas de Caja Blanca: se obtuvo en la primera iteración 8 defectos, 5 pertenecen a Error en variable lógica y 3 se clasifican de tipo Variables de bucles modificadas de forma inapropiada.
En los tres tipos de pruebas, se corrigieron los defectos detectados en la primera iteración, lo que trajo consigo que en la segunda iteración no se detectaran defectos en ninguna de las pruebas ejecutadas, demostrando así la calidad de los componentes propuestos y, a su vez, el cumplimiento de los requisitos del sistema.
Hasta el momento se cuenta con las funciones previamente expuestas, las cuales conforman en su conjunto a BioBlender, permitiendo su utilización en las más recientes versiones de Blender, basadas en versiones de Python vigentes hasta la fecha. Dicho resultado, contribuye al continuo desarrollo de este paquete de software, posibilitando la creación e implementación de futuras funciones que se sumen a la interpretación en el espacio tridimensional de propiedades de superficie como el MLP y el EP, importantes para la comprensión del com- portamiento de las moléculas con su entorno y que realicen aportes en el escenario actual, donde el avance de las tecnologías biológicas es fundamental en esta era, marcada de muertes y enfermedades provocadas por una gran variedad de patógenos.
CONCLUSIONES
La evidencia presentada en conjunto con los resultados de las pruebas de software realizadas, ayudaron a de- mostrar y evaluar la calidad de cada uno de los componentes que conforman BioBlender, demostrando de esta forma, su funcionamiento en versiones actuales de Blender, cumpliendo los objetivos trazados en la investigación. Dicho esto, es posible representar en el espacio tridimensional biomoléculas utilizando una versión vigente de la plataforma y obtener de ellas propiedades de superficie como el potencial lipofílico molecular y el potencial electrostático, brindando una mejor comprensión de información bioquímica importante, como la hidropatía o las cargas. La migración del código a Python v3.7.5 en adelante, un paso importante para lograr la reincorporación del módulo a Blender, constituye, a su vez, la base para futuras implementaciones, con el objetivo de ampliar las expectativas que se tienen con él.
La investigación no se detiene en estos resultados, ya que la plataforma en la que está desarrollado este paquete de software se encuentra en constante movimiento, lo que implica el estudio de ella continuamente para mantenerse al ritmo de las nuevas tecnologías de animación 3D, haciéndolo un proceso a largo plazo. Su dependencia a Blender, hace que sea imprescindible el surgimiento de versiones futuras de BioBlender, por lo que aún se trabaja en ello, buscando optimizar el trabajo realizado y buscando nuevas vías para la realización de futuras funciones.

