










 Serviços customizados
Serviços customizados Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroducción
La industria biofarmacéutica cubana produce, aproximadamente, el 87 % de los medicamentos de consumo de la población cubana, según el Cuadro Básico de Medicamentos (CBM) de 2014 1, lo cual depende de la importación de materias primas, materiales de envase y una parte de los productos terminados.
El Paracetamol Solución Oral 24 mg/mL, es un medicamento incluido en el Cuadro Básico de Medicamentos (CBM) del 2014 1, que es producido por la empresa Laboratorio Farmacéutico Líquidos Orales de Bayamo, Medilip. El paracetamol o acetaminofén, pertenece al grupo farmacológico de los analgésicos no opioides 2, y como sustancias mediadoras del dolor, es ampliamente utilizado como analgésico y antipirético. Este fármaco se administra por vía oral e intravenosa, siendo comercializado en varias formas farmacéuticas como tabletas, gotas, soluciones e inyectables.3
En los años 2017 y 2018, se decidió desarrollar por parte del grupo de Investigación y Desarrollo de Medicamentos (I+D) de Medilip, una nueva formulación de solución oral, incluyendo el par metilparabeno (MPB) y propilparabeno (PPB), para mejorar las características organolépticas y la estabilidad microbiológica de la misma, por lo que se hizo necesario el desarrollo de una nueva técnica analítica encaminada a la determinación cuantitativa simultánea de acetaminofén (AAF), metilparabeno (MPB) y propilparabeno (PPB) en Paracetamol Solución Oral 24 mg/ mL.
En la actualidad, para la competitividad dentro de la industria farmacéutica, se presta especial atención a los temas relacionados con la calidad y seguridad de los productos farmacéuticos, estando convencidos, que estos son vitales para la asistencia a la salud. Además, todas las partes que intervienen en la producción de medicamentos deben velar por un total cumplimiento de las normativas nacionales e internacionales que satisfacen las demandas y leyes que rigen la comercialización y registro en cada país, en particular, y para la exportación de manera general. Por esta razón, constantemente se busca implementar nuevas tecnologías y formulaciones farmacéuticas, con el fin de lograr sistemas rigurosos de calidad para proporcionar y garantizar a los consumidores un producto de alta calidad que cumpla con las especificaciones establecidas.
La USP-414 establece, que se deben aplicar diferentes esquemas de validación, por la lógica razón que existen diferentes procedimientos de ensayos. Esta farmacopea clasifica en varias categorías los métodos analíticos para su validación. El método objeto de estudio de esta investigación, se clasifica como procedimientos analíticos para la cuantificación de componentes mayoritarios de materias primas o ingredientes activos (incluyendo preservos) en productos farmacéuticos terminados. Los parámetros de validación que hay que tener en cuenta, pueden variar en dependencia de los requisitos legales exigidos por las diferentes organizaciones.
El objetivo del presente trabajo es validar exhaustivamente el nuevo método analítico, mediante los ensayos definidos por la resolución 40/2014 “Validación de métodos analíticos”, del Centro para el Control Estatal de la Calidad de los Medicamentos y Dispositivos Médicos (CECMED) 5 y la Norma Cubana NC TS 368:2010, Guía para la validación de métodos de ensayo químicos.6 Para demostrar la confiabilidad del método cromatográfico en la determinación simultánea del ingrediente farmacéutico activo (AAF) y los preservos, se realizaron pruebas preliminares como idoneidad del sistema y selectividad. La validación se completó con pruebas de linealidad, exactitud, precisión, robustez y límites de detección y cuantificación, acorde con las normas nacionales e internacionales.7,8,9
Materiales y métodos
Equipos, materiales y reactivos
Los equipos utilizados para el desarrollo del método fueron debidamente calibrados y validados; así como, la cristalería empleada (clase A) fue verificada por la Oficina Territorial de Normalización (OTN) e instituciones correspondientes.
Equipos
Cromatógrafo líquido de alta eficacia con detector de longitud de onda variable, HPLC KONIK 580/España y HPLC AZURA/Alemania
Balanza analítica digital, 250 g/d= 0,01 mg, Sartorius/España
Baño ultrasónico BANDELIN/Alemania
Materiales
Matraces de 10 mL y de 20 mL, clase A, Pobel/España
Micropipeta de 10 µL-5000 µL, ITELTA/España
Pipeta graduada de 1 mL, clase A, Pobel/España
Microjeringuilla, punta 3, de 100 µL
Filtros Ministart RC 4 de 0,45 μm
Embudo de cristal pequeño
Vaso de precipitado de 100 mL
Reactivos
Método empleado
El método para la cuantificación de AAF, MPB y PPB en lotes de Paracetamol Solución Oral, se basa en la separación por HPLC de los principios activos de interés de sus impurezas y otras posibles interferencias, utilizando para ello una columna C8, mediante detector UV-VIS de longitud de onda variable. Se empleó el método del patrón externo. Las lecturas fueron editadas sobre plataforma Clarity. Konik Chrom Plus 2005. Para los cálculos y el análisis estadísticos se utilizaron los programas Statgraphics Plus Centurion 18 Professional, versión 18.1.12 (64 bit) y Microsoft® Office Excel 2019. Teniendo en cuenta las similitudes en las estructuras, propiedades físicas y espectros de absorción en la zona de ultravioleta-visible (UV/vis), se escogió la longitud de onda igual a 258 nm.10,11,12
Preparación de la disolución de muestra y de referencia
Se prepararon por disoluciones sucesivas, hasta lograr las muestras de análisis de 120 µg/mL (AAF), 9 µg/mL (MPB) y 1 µg/mL (PPB). La primera disolución se arrastró, cuantitativamente, con acetonitrilo (CH3CN), y el resto de las disoluciones con fase móvil (FM). Se destiló a través de filtro de poración 0,45 µm.
Preparación de la disolución de la muestra de paracetamol y parabenos para el estudio de los rangos
Hubo un procedimiento semejante al anterior, pero se logró concentraciones aproximadas como se muestran:
AAF: 96 µg/mL (80 %), 108 µg/mL (90 %), 120 µg/mL (100 %), 132 µg/mL (110 %) y 144 µg/mL (120 %)
MPB: 7,2 µg/mL (80 %), 8,1 µg/mL (90 %), 9,0 µg/mL (100 %), 9,9 µg/mL (110 %) y 10,8 µg/mL (120 %)
MPB: 0,8 µg/mL (80 %), 0,9 µg/mL (90 %), 1,0 µg/mL (100 %), 1,1 µg/mL (110 %) y 1,2 µg/mL (120 %)
Cálculos
Se empleó la fórmula dada por la ecuación (1), para calcular la concentración de los analitos:

donde:
concentración de los analitos en la muestra ( )
concentración de las referencias en ( )
área bajo la curva de los picos correspondientes a los analitos en la muestras ( )
área bajo la curva de los picos correspondientes a los analitos de referencia ( )
factor de dilución de la muestra (200)
Parámetros de validación4-6,8,13
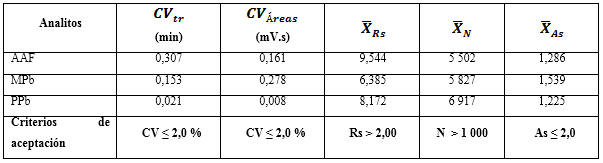
La idoneidad del sistema cromatográfico se evaluó a través de las respuestas de cinco réplicas de los picos de la disolución de referencia del paracetamol y los parabenos. Se determinaron las variabilidades de los parámetros cromatográficos: áreas de los picos (Am), platos teóricos (N), la resolución (Rs) y factor de cola o asimetría (As). Para el primero, el coeficiente de variación debe ser menor o igual a 2,0 % (CV ≤ 2,0 %). La eficiencia de la columna, no debe ser mayor de 1 000 platos teóricos (N ≥ 1 000); la Rs debe ser mayor de 2,0 (Rs ≥ 2,0), y el promedio de As no debe exceder 2,0 (As ≤ 2,0 %).
Selectividad: se realizaron las corridas correspondientes al placebo y solución de la muestra sometida a hidrólisis básica , hidrólisis ácida y calor, todas a reflujo durante una hora. Además, se realizó una oxidación sobre 1 mL de la muestra con tres gotas de , y para la fotosensibilidad se aplicó luz ultravioleta a una longitud de onda (λ) igual a 254 nm (con lámpara UV KAMAK, de procedencia inglesa). Se determinaron las posibles interferencias como criterio de selectividad del método.14
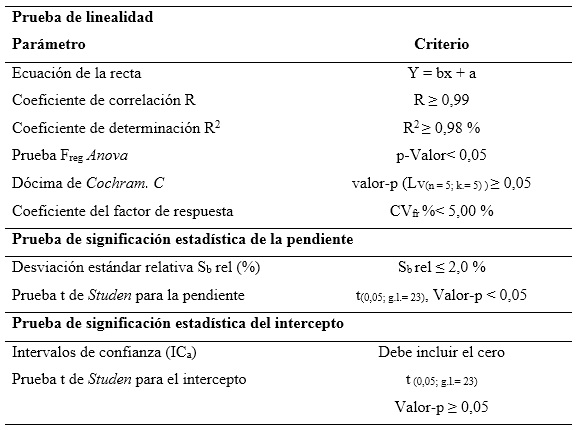
Linealidad: se realizó una curva de calibración en el intervalo de concentraciones comprendido entre 97,2 µg/mL y 145,8 µg/mL para el AAF; entre 7,296 µg/mL y 10,944 µg/mL para el MPB, y entre 0,82 µg/mL y 1,23 µg/mL para el PPB; correspondiente al intervalo de 80 a 120 % de las concentraciones nominales. El análisis se realizó por quintuplicado. Se determinó la ecuación de la recta, el coeficiente de correlación (≥ 0,990), el coeficiente de determinación (> 0,98) y el CV de los factores de respuesta (≤ 5 %). Se evaluó la varianza de la pendiente de la línea de regresión, desviación estándar, desviación estándar relativa, límites de confianza de la pendiente y grado de significación; además, se verificó la proporcionalidad.15-18 Todos los datos obtenidos fueron procesados con los softwares Microsoft Excel 2019 y el paquete profesional STATGRAPHICS CENTURION 18. Los criterios de aceptación aparecen en la tabla 1.
Exactitud del método: se empleó el método de recuperación, preparando placebos cargados con AAF, MPP, PPB. Se ejecutó con utilización de materiales de referencias certificados preparados en el laboratorio (materiales de referencia químico, MRQ) con un contenido conocido del analito. Se realizó el modelo de cinco réplicas para tres concentraciones diferentes: 80, 100 y 120 %. Se determinaron los por cientos de recuperación (% Recup), la desviación estándar (S), varianzas (S2), en cada nivel y el coeficiente de variación global para cada uno de los analitos analizados: AAF, MPP, PPB. Se aplicó la prueba de Cochran y la prueba t de Student de una cola para la comparación estadística del % Recup respecto al 100 % y su homocedasticidad. Los criterios de aceptación para exactitud se establecieron según normas de referencia.5 Además, se determinaron los intervalos de confianzas.6,13
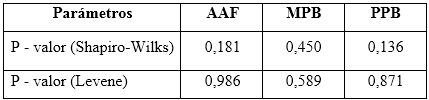
Presión del método: para desarrollar este estudio se evaluaron las precisiones en condiciones de repetibilidad e intermedia. Para estimar la precisión, se analizaron por seis analistas, seis réplicas de una muestra del mismo material al 100 % de la concentración nominal, durante tres días. A los resultados se les realizaron pruebas de normalidad a través del estadígrafo Shapiro-Wilks y gráficos de frecuencia (histogramas), las verificaciones de varianzas se infirieron con la prueba de Levene. Como criterio de cada prueba, se estimaron los valores de probabilidad de cada dócima, los que deben ser superiores a 0,05.6 El procesamiento estadístico se llevó a cabo con el paquete profesional Statgraphics Centurion XV, versión 15.2.14
Precisión en repetibilidad: se determinaron las medias, desviaciones estándares típicas y el coeficiente de variación CV % (CV ≤ 1,50), respecto al analista. Los datos para el procesamiento estadístico de obtuvieron de los análisis de varianzas correspondientes.5 Se reportan, además, los intervalos de confianza para la repetibilidad.19
Precisión intermedia: se continuó el diseño propuesto. Se determinaron las desviaciones típicas de la repetibilidad del método (sr) y la desviación típica de la reproducibilidad (sR), calculados a partir de los resultados de los cuadrados medios de los modelos de ANOVAs, y mediante las fórmulas (2), (3) y (4) descritas a continuación.5,6


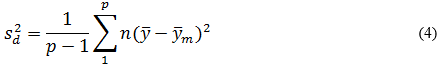
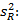
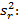
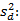
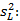
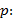
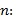
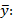
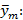 donde varianza de la reproducibilidad interna varianza de la repetibilidad (cuadrado medio dentro de grupos) varianza media (cuadrado medio entre de grupos) varianza entre días y analistas número de analistas números de réplicas por analistas media por analista Media de todos los analistas
donde varianza de la reproducibilidad interna varianza de la repetibilidad (cuadrado medio dentro de grupos) varianza media (cuadrado medio entre de grupos) varianza entre días y analistas número de analistas números de réplicas por analistas media por analista Media de todos los analistasFueron utilizadas las muestras al 100 % fueron preparadas mediante dilución. Como criterio de aceptación, los CVR% no deben superar el 1,50 %.
Límites de detección y de cuantificación: para este estudio se utilizó el método de la relación señal/ruido a partir de las curvas de linealidad obtenidas en el ensayo de linealidad, propuesto por EURACHEM20,21. Las evaluaciones de los límites de detección (LDD) y de los límites de cuantificación (LDC) se realizaron por determinación de señal de ruido del blanco por extrapolación (Ybl) y las desviaciones del blanco (Sbl) a partir de la extrapolación de la curva de las desviaciones estándares de los cincos niveles de los estudios de la linealidad. Se tuvo en cuenta el criterio de relación señal/ruido para el LDD mayor que tres, y para el LDC mayor que diez. Se analizaron los parámetros respuestas de los blancos (Ybl); se establecieron las ecuaciones de las rectas con sus respectivos coeficientes de determinación (r2 ≥ 98,0 %), desviación estándar del blanco (Sbl), a partir de diez determinaciones de un blanco, y se calcularon los límites de detección cuantificación según la ecuación (5):6
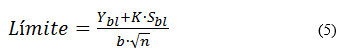
donde
K: factor matemático para la determinación de los límites de detección (3) y de cuantificación (10)
b: pendiente de la recta
n: número de determinaciones
A partir de los LDCs se realizaron pruebas de disoluciones de los parabenos y paracetamol a los niveles de cuantificación. Se preparó una disolución a niveles de concentración de LDC. Se realizaron diez determinaciones, y a los resultados se les ejecutaron pruebas de exactitud por recuperación. Se reportaron las recuperaciones medias con la prueba de significación de t de Student, para un valor de probabilidad superior a 0,05. La precisión se comprobó por la determinación del coeficiente de variación en por ciento. Los criterios de aceptación se establecieron de acuerdo con los propuesto por la FDA, según el nivel de concentración determinado.22
Robustez: el diseño factorial Plackett-Burman, con la variante Youden-Steiner6,23 fue el utilizado para evaluar la robustez con cambio a un plan factorial fraccionado. Para este diseño se propuso analizar tres factores, con cuatro experimentos, y como variable de respuesta se empleó la concentración en por ciento para cada uno de los analitos. Se tuvieron en cuenta los factores experimentales como el tipo de relleno de la columna (esférico e irregular), variación de la longitud de onda y el equipo HPLC-UV/vis. Las condiciones nominales, así como las variaciones de los tres factores analizados, se pueden observar en la tabla 2.
Tabla 2 Combinación de condiciones analizadas en el ensayo de robustez, según el diseño factorial optimizado
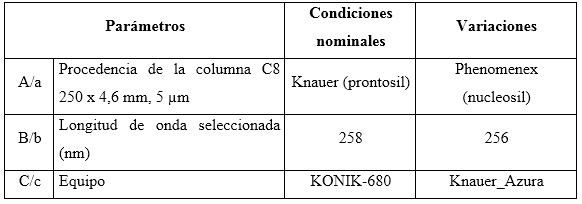
El diseño factorial de los resultados se lleva acabo según se muestra en la tabla 3.
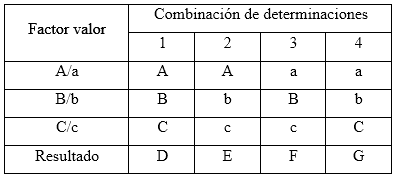
Las variaciones de los experimentos (DA, DB y DC), se calcularon como indica el diseño experimental, sobre la base de la diferencia entre los resultados de los factores en los niveles alto y bajo, según las ecuaciones descritas a continuación:
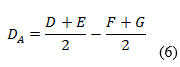
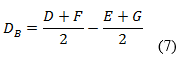
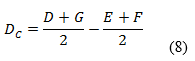
Como criterio de aceptación se sugiere, que no deben exceder a dos veces la desviación típica del estudio de precisión en repetibilidad (2sr). Una diferencia mayor (2sr) significa, que el efecto del factor es estadísticamente significativo al nivel de confianza de 95 %.
Resultados y discusión
La idoneidad del sistema cromatográfico
En la tabla 4 se resumen las respuestas de los parámetros cromatográficos, luego de la aplicación del método en el análisis al AAF y los dos parabenos. Los coeficientes de variación de los tiempos de retención (CVtr) y las áreas (CVÁreas) de las réplicas inyectadas, rindieron valores por debajo de 2,0 %. Para las variables resolución (Rs), platos teóricos (N) y asimetría (As), se lograron resultados dentro de los criterios de aceptación, mostrando poca variabilidad entre las réplicas; por lo que se demuestra que el sistema cromatográfico empleado funciona correctamente.20
Selectividad
Al comparar los cromatogramas de las corridas de muestra y placebo se observa, que en la formulación no existen interferencias que afecten la calidad del método propuesto. Los cromatogramas se exponen en la figura 1. Iguales observaciones se obtienen con las corridas de las condiciones estresantes bajo la acción de la luz ultravioleta y el calor, que no muestran interferencias significativas a la calidad del sistema.
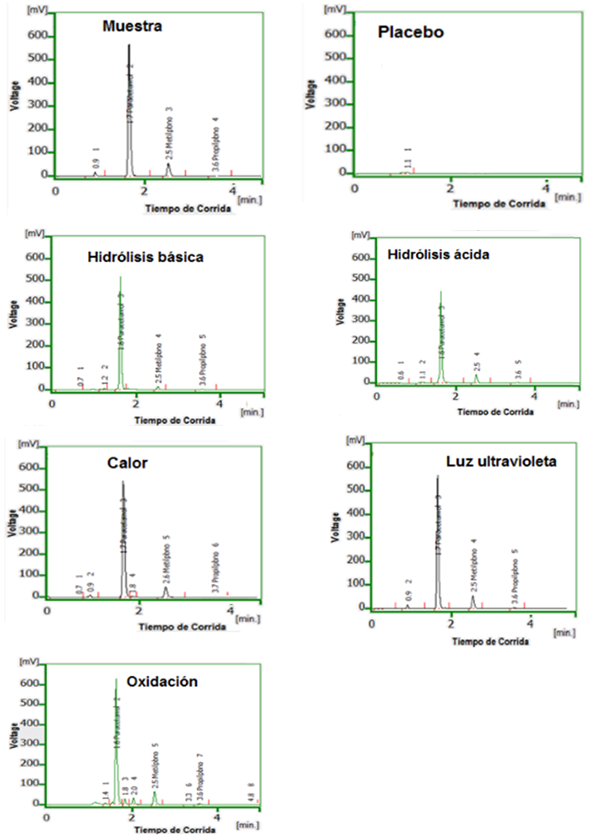
Fig. 1 Cromatogramas del estudio de selectividad para acetaminofén (1,7 min), metilparabeno (2,5 min) y propilparabeno (3,6 min)
Los productos de degradación de las hidrólisis ácida y básica tampoco interfieren, ya que al formarse sustancias de menores masas y volumen atómicos, son menos retenidos, eluyendo en tiempos de retenciones diferentes. Por último, en el caso de la oxidación que muestra mayor número de interferencias, debido a p-aminofenol, p-nitrofenol, anilina (paracetamol) y los compuestos fenólicos de los parabenos (ácido p-hidroxibenzoico)8, no superponen a los tiempos de retención de los analitos de interés. Como conclusión de los ensayos realizados y las respuestas obtenidas a las condiciones estresantes de degradación, se considera que el método analítico que se propone cumple con el ensayo de selectividad (especificidad farmacéutica).
Linealidad
La tabla 5 muestra el resumen de los resultados del estudio de linealidad para el AAF, MPB y PPB en el intervalo de 80 a 120 por ciento, respecto a las concentraciones nominales en la formulación farmacéutica (de 96 a 144 µg/mL para el AAF; 7,2 a 10,8 µg/mL para el MPB y 0,8 a 1,2 µg/mL para el PPB).
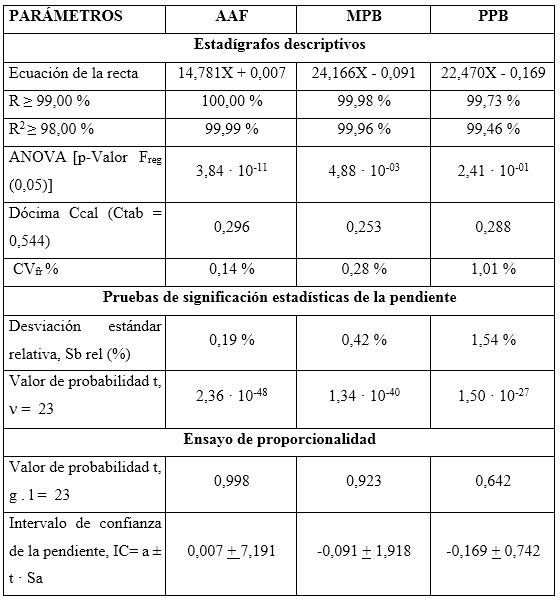
Los coeficientes de correlación y determinación indicaron, que los modelos matemáticos propuestos muestran correlaciones superiores al 99,0 %, y explican más del 98,0 % de las desviaciones en las determinaciones para cada nivel. Los análisis de varianzas de un solo factor demostraron la fuerte relación seriada entre las variables dependiente (concentración) e independiente (áreas bajo la curva), obteniendo valores de probabilidad por debajo de 0,05. La prueba de homocedasticidad aplicada a través de la prueba C de Cochran arrojó, con una certeza de 95 % de confianza, que las variabilidades de cada nivel se consideran iguales estadísticamente. Los coeficientes de variación de los factores de respuestas, estuvieron todos por debajo de 5,0 %, por lo que se estima una linealidad adecuada. Se obtuvieron las ecuaciones lineales, a las que se les aplicaron las pruebas de significación estadística de las pendientes y los interceptos.
Los cálculos de las desviaciones estándares de las pendientes indican que las variaciones de los residuales se encuentran dentro de los límites de alerta y de acción, con valores inferiores al 2,0 %, mientras que los valores de probabilidades inferiores a 0,05 sugieren relaciones lineales adecuadas. Las determinaciones de los intervalos de confianzas de los interceptos incluyen el cero. Las gráficas de los ajustes por mínimo cuadrado, que se muestran en la figura 2, evidencian las distribuciones aleatorias de residuales en torno a cero, lo que confirma la linealidad.
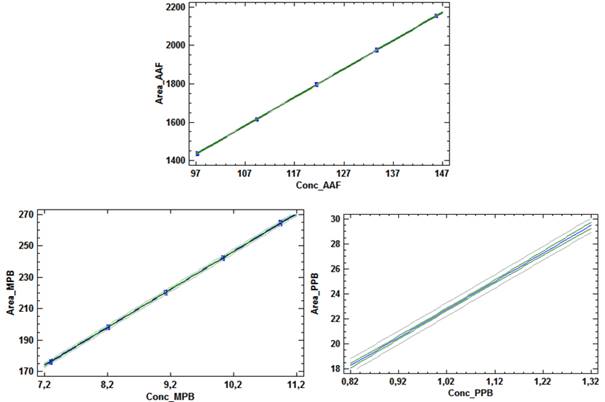
Se concluye, que se puede considerar que las relaciones entre las concentraciones y las respuestas del método propuesto se pueden considerar lineales en el intervalo de concentraciones de 80,0 a 120,0 %, respecto a los valores nominales.
Exactitud del método
Los resultados del estudio de homocedasticidad mostraron (ver tabla 6), que las varianzas se pueden considerar constantes para los niveles estudiados en cada uno de los analitos. Las homogeneidades de las varianzas se infirieron a través de los resultados de las dócimas de Cochran al obtenerse valores calculados inferiores al valor crítico de 0,778 (K = 3, n = 5).
Tabla 6 Resultados estadísticos del estudio de exactitud para acetaminofén (AAF), metilparabeno (MPB) y propilparabeno (PPB)
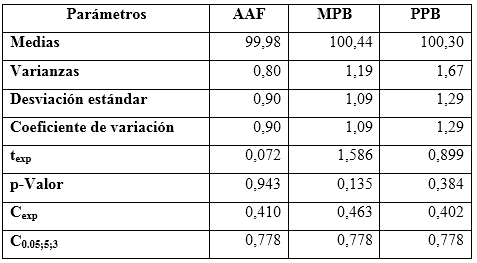
Los porcientos de recuperaciones obtenidos no presentaron diferencias estadísticas significativas, respecto al 100,0 % de recuperación teórico, corroborado por el estadígrafo de t de Student, mostrando valores de probabilidad superiores al 0,05.5
En la tabla 7 se observan los valores medios con sus intervalos de confianzas, que posibilitan la determinación de los sesgos para cada analito en cuestión.
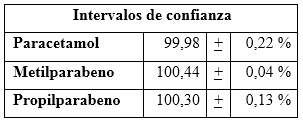
Precisión del método
Los resultados demostraron que en los tres casos se cumple con distribuciones normales, según la prueba de Shapiro-Wilks, al obtenerse valores de probabilidades superiores a 0,05. Las homogeneidades de las varianzas fueron verificadas a través del estadígrafo de Levene. Se concluyó, que las dispersiones de los resultados para las concentraciones de todos los analitos de interés en el estudio de condiciones intermedias, respecto a los días analizados y entres los analistas, se consideran homogéneas. Los valores obtenidos para el estudio de la precisión se reflejan en la tabla 8.
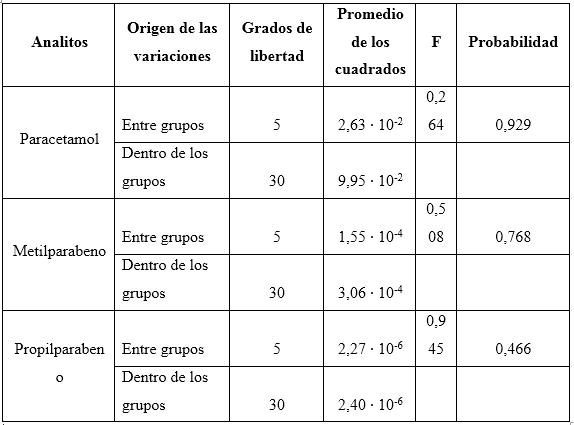
Los valores de probabilidad de la prueba de ANOVA (tabla 9), en todos los casos estudiados indican que los valores de las concentraciones, comparados respecto a las dos condiciones, se pueden consideras iguales, al obtenerse valores superiores al 5 %, para un nivel fiducial de 95,0 %.
Precisión en repetibilidad
En la tabla 10 se observan los resultados de los estadígrafos descriptivos. Los valores de los coeficientes de variación expresados en por ciento, indican que las variabilidades no sobrepasan el 1,50 % de los resultados, lo que hace considerar que el método es preciso en condiciones de repetibilidad, para un 95 % de confianza. En la determinación de los resultados se tuvo en cuenta los grados de libertad (30 para los analistas, y el valor de la t de Student para un 95 % de probabilidad (2,03). Los decimales en cada caso fueron expresados hasta la última cifra significativa.
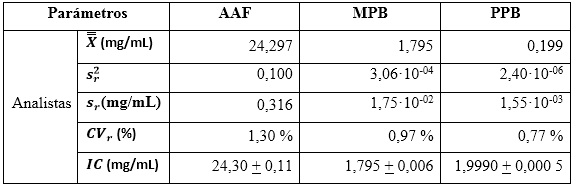
Precisión intermedia
A partir de la ANOVA, se calcularon las varianzas combinadas entre días y analista 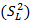 , las
, las 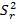 del estudio de repetibilidad y varianza media
del estudio de repetibilidad y varianza media 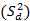 , que corresponde a los valores de los cuadrados medios dentro de grupos, (como se indican en las ecuaciones 2, 3 y 4) y se determinaron las varianzas de la reproducibilidad . Los intervalos de confianzas observados se determinaron con los valores de grados de libertad 6, como resultado total, y un valor para la t de Student correspondiente a 2,03. Los coeficientes de variación, en ningunos de los casos, muestran valores superiores al criterio establecido, lo que indica que los errores aleatorios no causaron impactos negativos significativos sobre los resultados. Las variaciones pueden justificarse estadísticamente con un nivel de confianza de 95,0 %. Los resultados se pueden analizar en la tabla 11.
, que corresponde a los valores de los cuadrados medios dentro de grupos, (como se indican en las ecuaciones 2, 3 y 4) y se determinaron las varianzas de la reproducibilidad . Los intervalos de confianzas observados se determinaron con los valores de grados de libertad 6, como resultado total, y un valor para la t de Student correspondiente a 2,03. Los coeficientes de variación, en ningunos de los casos, muestran valores superiores al criterio establecido, lo que indica que los errores aleatorios no causaron impactos negativos significativos sobre los resultados. Las variaciones pueden justificarse estadísticamente con un nivel de confianza de 95,0 %. Los resultados se pueden analizar en la tabla 11.
Tabla 11 Estadígrafos descriptivos para el estudio de la precisión intermedia e intervalos de confianzas
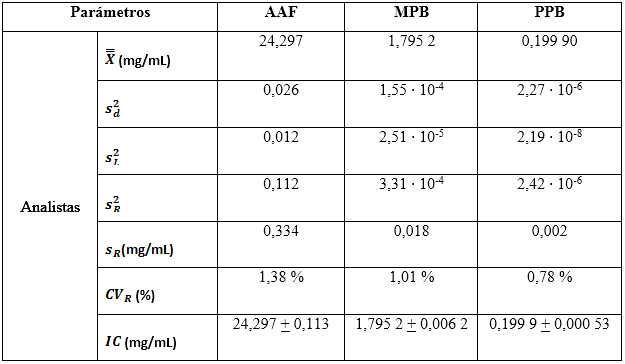
Este tipo de diseño puede proporcionar una forma eficaz de obtener suficientes grados de libertad, para las estimaciones de la repetibilidad y precisión entre los grupos. Al analizar el diseño propuesto, seis analistas de seis repeticiones conducen a 30? y 35? de libertad para la estimación de la repetibilidad y precisión entre series, respectivamente.6,20
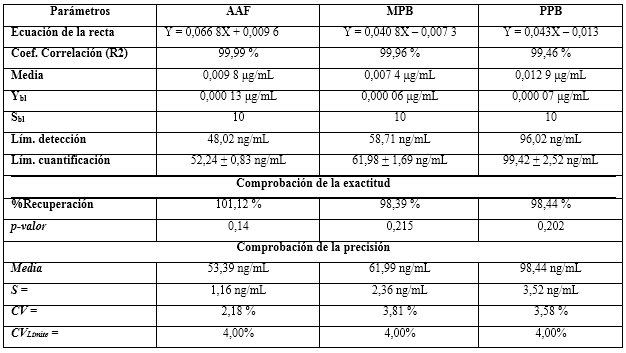
Límites de detección y de cuantificación
Los límites de cuantificación quedaron en el orden de 50 a 100 ng/mL, por lo que se determinó como valor límite para los coeficientes de variaciones hasta un 4,0 %, no siendo excedido en ninguno de los casos. El ensayo de recuperación infirió la no existencia de diferencias estadísticas significativas entre los por cientos recuperados y el 100,00 %, justificados con los p-valores superiores a 0,05 en todos los casos. Los resultados se resumen en la tabla 12. Las pruebas de exactitud y precisión resultaron adecuadas para los niveles de cuantificación.20 Los valores obtenidos por el método que se propone, resultaron más sensibles y adecuados para ser utilizados en estudios de cuantificación de trazas y validación de limpieza.
Robustez
El resultado del estudio de la robustez se muestra en la tabla 13. Los valores obtenidos de las dispersiones calculados para cada factor se compararon con el doble de la desviación estándar de la repetibilidad observándose que en ninguno de los casos excede el valor tomado como límite. Esto indica que el método es robusto para la cuantificación de los analitos de interés.
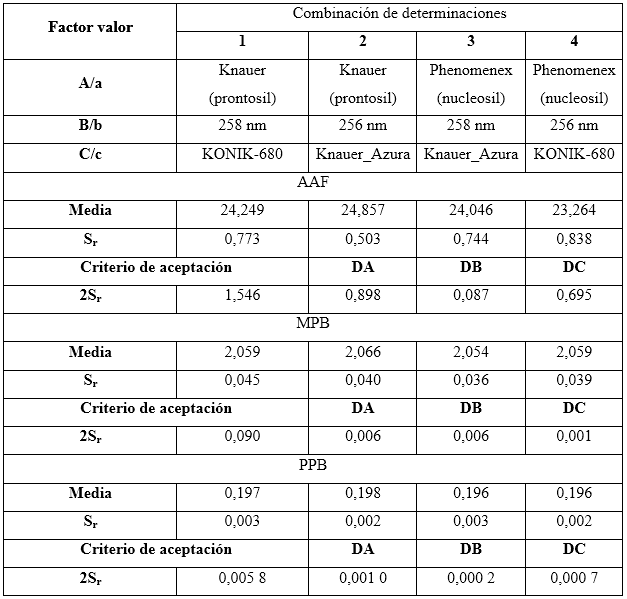
El método es capaz de producir resultados aceptables ante desviaciones menores del método, por lo que resulta un indicador confiable para continuar con la validación del método.
Conclusiones
El método de ensayo para la determinación de los contenidos de acetaminofén, metilparabeno y propilparabeno por cromatografía líquida de alta eficacia, fase inversa, utilizado para el control de la calidad de formulaciones de Paracetamol Solución Oral 120 mg/5 mL y para su estudio de estabilidad, resultó ser específico, preciso, exacto y lineal en el intervalo de concentraciones de 96 a 144 µg/mL para el AAF; 7,2 a 10,8 µg/mL para el MPB, y 0,8 a 1,2 µg/mL para el PPB. Los límites de detección determinados fueron de 48,02 ng/mL; 58,71 ng/mL; 96,02 ng/mL, y de cuantificación de 52,24 + 0,83 ng/mL; 61,98 + 1,69 ng/mL; 99,42 + 2,52 ng/mL para AAF, MPB, PPB, respectivamente.

