










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Farmacia
versión On-line ISSN 1561-2988
Rev Cubana Farm v.43 n.1 Ciudad de la Habana ene.-abr. 2009
ARTÍCULOS ORIGINALES
Bioequivalencia de una formulación cubana de carbamazepina con el producto líder
Bioequivalent of a Cuban formula of Carbamazepine with the leader product
Alejandro Saúl Padrón YaquisI; Narda María Jiménez AlemánII; Lic. Jorge Ernesto Calero CarbonellII; Carlos A. González DelgadoIII; Armando Correa FernándezIV; Lourdes Olivera LluanoV; Alina Díaz MachadoVI
IDoctor en Ciencias Farmacéuticas. Ingeniero en Tecnologías Químicas Nucleares. Master en Ciencias Técnicas. Investigador Auxiliar. Centro de Investigación y Desarrollo de Medicamentos (CIDEM). La Habana, Cuba.
IILicenciado en Ciencias Farmacéuticas. Aspirante a Investigador. CIDEM. La Habana, Cuba.
IIIMaster en Farmacología. Investigador Auxiliar. Centro Nacional de Toxicología (CENATOX). La Habana, Cuba.
IVLicenciado en Química. CENATOX. La Habana, Cuba.
VLicenciada en Química. Master en Química Analítica. Investigadora Agregada. CENATOX. La Habana, Cuba.
VIResidente de Farmacología. CENATOX. La Habana, Cuba.
RESUMEN
Se demostró la intercambiabilidad terapéutica mediante un estudio de bioequivalencia en 25 voluntarios sanos de una formulación cubana de carbamazepina de 200 mg con respecto al producto innovador (Tegretol®), en condiciones de administración a dosis única; el diseño experimental fue cruzado a doble ciegas y aleatorizado; el periodo de lavado fue de 15 días. Las extracciones para la obtención del plasma, se realizaron a las 0; 0,5; 1; 1,5; 2; 3; 4; 6; 8; 10; 12; 16; 20; 24; 48; 72; 96 y 120 h. Para el análisis se empleó un método analítico por cromatografía líquida de alta eficiencia, isocrático en fase reversa con detección ultravioleta. Se estimaron mediante técnicas "no compartimentales" los parámetros farmacocinéticos (AUC0-t, AUCt-¥, Cmax, Tmax y T1/2) representativos de la biodisponibilidad en magnitud y velocidad. Sobre la base de los resultados estadísticos, ambas formulaciones resultaron bioequivalentes.
Palablas clave: Carbamazepina, bioequivalencia, Tegretol.
ABSTRACT
It was possible to demonstrate the therapeutic interchange level by means of a bioequivalence study in 25 healthy volunteers of a Cuban formula of Carbamazepine(200 mg) according to the innovative product (Tegretol®) for a administration of unique dose; the experimental design was of crossed, double-blind and random type; washing period was of 15 days. Extractions for plasma obtaining were performed at 0, 0,5, 1, 1,5, 2, 3, 4, 6, 8, 10, 12, 16, 20, 24, 48, 72, 96 and 120 hours. In analysis we used an analytical method by high performance liquid chromatography, isocratic type in reverse phase with UV detection. By means of "non-compartment techniques" pharmacokinetic parameters (AUC0-t, AUC t-¥, Cmax, Tmax, and T½) were estimated, which are representative of bioavailability in magnitude and speed. On the base of statistical results, both formulae were bioequivalent.
Key words: Carbamazepine, bioequivalence, Tegretol.
INTRODUCCIÓN
El desarrollo de nuevas formulaciones genéricas de medicamentos necesita que se demuestre la intercambiabilidad terapéutica, para ello se requiere en ocasiones la realización de estudios de bioequivalencia. Bioequivalencia significa la ausencia de una diferencia significativa en la tasa y el grado en que el principio activo o la fracción activa en equivalentes farmacéuticos o alternativas farmacéuticas está disponible en el sitio de acción del fármaco cuando se administra a la misma dosis molar en condiciones similares en un estudio apropiadamente diseñado.
En el presente trabajo se demostró la bioequivalencia en voluntarios sanos cubanos de una formulación de carbamazepina 200 mg con respecto al producto innovador, Tegretol®, bajo condiciones de ayuno con administración a dosis única.
La carbamazepina es un fármaco de primera línea en el tratamiento de las epilepsias parciales simples y complejas, de gran uso en pacientes con epilepsia tónico-clónica generalizada y en el tratamiento de las neuralgias del trigémino y del glosofaríngeo.1,2 Su farmacocinética se ha estudiado de forma amplia, demostrándose variaciones biológicas individuales apreciables en su absorción y biotransformación; las tabletas de carbamazepina elaboradas por distintos laboratorios farmacéuticos presentan a menudo variaciones en su biodisponibilidad.3 En la literatura médica se han comunicado diferentes casos de pacientes tratados con la formulación de referencia (Tegretol®) que han sufrido cambios de concentración plasmática acompañados de cambios en la eficacia o toxicidad del fármaco cuando se les cambió de Tegretol® por una formulación genérica de carbamazepina.4-10
En el humano, la carbamazepina se absorbe relativamente despacio y de manera errática desde los comprimidos, se distribuye con rapidez por todos los órganos y tejidos, no presenta afinidad por ningún órgano en específico, se fija a las proteínas plasmáticas en un 70-80 % y se metaboliza en el hígado, donde la vía epóxido de biotransformación es la más importante, dando lugar al metabolito activo denominado carbamazepina 10-11epóxido (carbamazepina 10-11 epóxido), el cual a su vez es hidratado transformándose en la transcarbamazepina-diol, que se excreta en la orina en forma libre y conjugada. La vida media de eliminación de la carbamazepina inalterada en la mayoría de los estudios de dosis única está entre 30 y 40 h con un valor promedio de 36 h.7-10
MÉTODOS
El estudio de bioequivalencia se realizó de acuerdo con el protocolo aprobado por el Comité de Ética del sitio clínico ubicado en el Centro Nacional de Toxicología (CENATOX) y el Centro para el Control Estatal de Medicamentos (CECMED) que autorizó el ensayo. Los productos en estudio fueron carbamazepina 200 mg ensayo 0501 de producción nacional (Laboratórios Medsol) y carbamazepina producto de referencia ensayo T4030 (Tegretol® Laboratorios Novartis). El diseño experimental fue cruzado a doble ciegas y aleatorizado, en el cual todos los individuos recibieron ambas formulaciones en 2 períodos, con un periodo de lavado de 15 días.
Participaron 25 voluntarios adultos sanos, de uno y otro sexos (18 hombres y 7 mujeres), con edad media de 35,2 ± 8,2 años (rango 22-50 años), el peso promedio fue de 66,25 ± 9,7 kg (rango 51-84 kg) y la talla media de 167,02 ± 10,4 cm (rango 151-184 cm). Estos no presentaron afectaciones significativas de tipo cardiaca, hepáticas, pulmonar, renal, neurológica, gastrointestinal o hematológica como se determinó por su historia clínica, el examen físico y exámenes de rutina del laboratorio (hematológicos, bioquímicos, orina y sexológicos).
A los voluntarios se les explicó de forma detallada las características y objetivos del estudio, así como las reacciones adversas que podrán presentarse a las dosis administradas y se recogió por escrito el consentimiento de cada voluntario en participar en el estudio, para lo cual firmaron el consentimiento informado según lo establecido en el acta de Helsinski y las Buenas Prácticas Clínicas.11-14
Los voluntarios ingresaron al sitio clínico a las 6:00 p.m. donde recibieron una comida estándar, las tabletas (referencia o prueba según lista aleatoria) se comenzaron a administrar entre 8 y 8:30 a.m., en ayunas (10 h) desde la noche anterior conjuntamente con un vaso de agua de 240 mL. Estos no ingirieron agua 1 h antes o después de la administración de las tabletas, permanecieron en reposo, no ingirieron alimentos hasta 4 h después de administrado el medicamento cuando se les administró un almuerzo estándar y no ingirieron bebidas carbonatadas ni con sustancias pertenecientes al grupo de las xantinas (café, té, cola, etc.), hasta que se tomó la última muestra de sangre.
Las extracciones de las muestras de sangre para la determinación de las concentraciones plasmáticas se realizaron a través de un catéter, el volumen de extracción fue de 5 mL. Las extracciones para la obtención del plasma se realizaron antes de administrar cada formulación (t= 0) y a las 0,5; 1; 1,5; 2; 3; 4; 6; 8; 10; 12; 16; 20; 24; 48; 72; 96 y 120 h. Una vez extraída la sangre las muestras fueron centrifugadas a 2 000 rev/min durante 10 min en una centrifuga MLWT-23. El plasma obtenido fue transferido a viales plásticos debidamente identificados, los cuales fueron almacenados a -20 oC hasta el momento de su análisis.
En viales Eppendorf de 1,5 mL se adicionó 500 µL de plasma al cual se le añadió1 mL de metanol previamente contaminado con estándar interno (propilparebeno, 0,5 µg/mL), se aplicó vortex y se centrifugó a 12 000 rev/min. Se tomó el sobrenadante y se inyectó (20 µL) en un sistema cromátográfico isocrático (fase móvil : mezcla acetonitrilo : hidrógeno fosfato de sodio pH= 3,3) en fase reversa (LiChrosorb RP-18, 10 µm, 250 x 4 mm) con detección ultravioleta (l-210 nm). El método analítico empleado fue desarrollado y validado para ese fin.15
Los parámetros farmacocinéticos representativos de la biodisponibilidad en magnitud y velocidad se estimaron mediante técnicas "no compartimentales".16 Se utilizó el programa de cálculo WinNonlin profesional, versión 2.1 A; Pharsight Co., 1998.
El tiempo de vida media plasmática (t½) se determinó a través de la relación 0,693/Ke, ajustando el intervalo de tiempo de la fase exponencial de eliminación terminal individual para cada sujeto. El área bajo la curva hasta el último tiempo de concentración plasmática cuantificable (AUC0-t), se calculó mediante el método de los trapezoides lineales. El área extrapolada (AUCt-¥) se calcula dividiendo el valor estimado de concentración para el último tiempo experimental (t) por el valor de la constante de eliminación terminal (Ke) y el área total como la suma del área experimental (AUC0-t) y el área calculada por extrapolación (AUCt-¥).17 Como parámetro para definir la bioequivalencia se calculó el área bajo la curva (AUC0-t) y la concentración máxima (Cmáx). Se estableció un coeficiente de variación de un 20 % con un nivel de significación (a) de 0,05, lo cual garantiza una potencia del 80 % para el AUC0 - t y la Cmáx.18
El análisis estadístico se realizó con el empleo las rutinas de cálculo del programa WinNonlin profesional, versión 2.1 A; Pharsight Co., 1998, se realizó un análisis de varianza (ANOVA) de doble vía y se descartó la presencia de los efectos de la formulación, periodo, secuencia y sujetos sobre el AUC0-t y la Cmáx en concordancia con el diseño cruzado del estudio.19
Se considera que la formulación de prueba es bioequivalente con respecto a la formulación de referencia si el intervalo de confianza del 90 % calculado según Westlake para el cociente de los promedios de los parámetros evaluados con un nivel de significación (a= 0,05) no sobrepasan el rango de bioequivalencia de 80 a 120 %, lo cual equivale al test Two-One Side de Shuirman.18
RESULTADOS
Ambas formulaciones fueron bien toleradas por los voluntarios; en la figura se muestran las medias (n= 25) de las concentraciones plasmáticas en cada tiempo para el medicamento de prueba y de referencia.

En la tabla 1 se resumen los parámetros farmacocinéticas y el tratamiento estadístico descriptivo tanto para el medicamento de referencia como de prueba.
Tabla 1. Estadística de las medidas farmacocinéticas de las medias de las concentraciones plasmáticas
del medicamento de referencia (r) y el de prueba (t) en los 25 voluntarios
| Parámetro | Tmax | Cmax | AUC0-t | (AUCt-∞) |
| Referencia | ||||
| Media | 8,560 | 5,929 | 327,510 | 398,197 |
| Desviación estándar | 1,960 | 0,973 | 43,185 | 74,766 |
| Error estándar | 0,392 | 0,195 | 8,637 | 14,953 |
| Varianza | 3,840 | 0,947 | 1864,982 | 5589,958 |
| Mínimo | 4,00 | 3,80 | 249,90 | 260,24 |
| Mediana | 10,00 | 5,92 | 322,74 | 399,77 |
| Máximo | 12,00 | 8,52 | 394,72 | 572,83 |
| Coeficiente de variación (%) | 22,9 | 16,4 | 13,2 | 18,8 |
| Límite inferior IC | 7,751 | 5,527 | 309,679 | 367,328 |
| Límite superior IC | 9,369 | 6,331 | 345,340 | 429,066 |
| Prueba | ||||
| Media | 7,120 | 5,789 | 331,876 | 393,386 |
| Desviación estándar | 2,804 | 1,368 | 77,631 | 104,902 |
| Error estándar | 0,561 | 0,274 | 15,526 | 20,980 |
| Varianza | 7,860 | 1,871 | 6026,503 | 11004,435 |
| Mínimo | 3,00 | 0,61 | 36,90 | 47,76 |
| Mediana | 8,00 | 6,15 | 344,85 | 402,88 |
| Máximo | 12,00 | 8,60 | 439,73 | 567,61 |
| Coeficiente de variación (%) | 39,4 | 23,6 | 23,4 | 26,7 |
| Límite inferior IC | 5,962 | 5,224 | 299,824 | 350,074 |
| Límite superior IC | 8,278 | 6,354 | 363,928 | 436,698 |
Tmax: tiempo en se alcanza la concentración plasmática máxima; Cmax: máxima concentración
plasmática; AUC0-t: área bajo de la curva hasta la última determinación; (AUCt-∞): área calculada
por extrapolación; IC: intervalo de confianza.
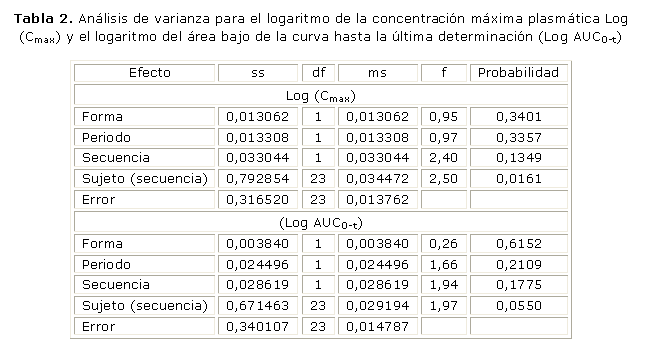
Los parámetros concentración máxima (Cmax) y área bajo la curva (el AUC0-t) cumplieron con los supuestos de normalidad en la distribución de las variables y homogeneidad de varianza. Se verificó que el efecto del periodo y de la formulación ambos fueron no significativos y se comprobó la inexistencia de efectos de residuos (carryovers). En la tabla 2 se presentan los resultados del análisis de varianza para estos parámetros.
En la tabla 3 se representan los resultados de los intervalos de confianza de las medidas farmacocinéticas empleadas para la evaluación de la bioequivalencia con un nivel de significación de 95,0 % (a=0,5).
DISCUSIÓN
Durante el estudio los voluntarios no presentaron incidencias de eventos adversos que provocaran la salida de este, lo que demuestra que ambas formulaciones son similares desde el punto de vista de su tolerancia.
Las curvas de las concentraciones plasmáticas medias en el tiempo para el medicamento de prueba y de referencia presentan perfiles muy similares y coinciden con el rango de concentraciones encontrados en otros estudios de carbamazepina administrada a esta misma dosis. De igual forma coinciden las medidas farmacocinéticas determinadas en el estudio con las reportadas por otros autores; la concentración máxima media osciló entre 3,8 y 8,5 µg/mL y 3,6 y 8,6 µg/mL para el medicamento de referencia y de prueba respectivamente. El tiempo en alcanzar la concentración plasmática máxima en los voluntarios varió entre las 3,0 y 12,0 h. El área bajo la curva fue similar en ambas formulaciones.
El análisis de varianza demostró que no existieron diferencias significativas en las diferentes fuentes de variación, formulación, periodo, sujeto y secuencia para la concentración plasmática máxima, y el área bajo la curva hasta el tiempo de la última determinación.
Los intervalos de confianza de 90,0 %, según Westlake para la razón de las medias del modelo paramétrico, satisfacen la condición de bioequivalencia para la concentración plasmática máxima, y el área bajo la curva hasta el tiempo de la última determinación.17 Se puede concluir que el medicamento prueba carbamazepina 200 mg de producción nacional es bioequivalente con el medicamento referencia carbamazepina (Tegretol®).
REFERENCIAS BIBLIOGRÁFICAS
1. Mc Namara, JO. Fármacos eficaces para el tratamiento de la epilepsia. En: Goodman-Gilman A. Las bases farmacológicas de la terapéutica. 9na ed. Mexico: Mc Graw-Hill Interamericana; 1996. p. 491-519.
2. Saavedra I, Passalacquar AR, Hernan Chavez G, Leandro Biagini A, Daniel Galdames P. Carbamazepina: Biodisponibilidad de cuatro productos farmacéuticos orales. Rev Med Chile. 1990; 118:1123- 8.
3. Gilman JF, Alvarez LA, Duchowry M. Carbamazepine toxicity resulting from generic substitution. Neurology. 1993; 43:2696-7.
4. Sachdeo RC, Belendiuk G. Generic versus branded carbamazepine. Lancet. 1987; 1:1432.
5. Diccionario de Especialidades Farmacéuticas. Edición No. 27, 1999. Editorial PLM. S.A. Tegretol, 1068.
6. Seppo Pynnonem. Pharmacokinetics of carbamazepine in man: A review. Ther Drug Monitoring. 1979; 1:409-31.
7. Bertilsson L, Torbjorm T. Clinical pharmacokinetics and pharmacological effects of carbamazepine and carbamazepine-10,11-Epoxide. Clin Pharmacikinetics. 1986; 11:177-198.
8. Levy RH. Pharmacokinetic of carbamazepine in normal man. Clin Pharmacol Ther. 1975; 17:657-68.
9. Westenberg HGM. Kinetics of carbamazepine and carbamazepine-epoxide determined by use plasma and saliva. Cin Pharmacol. 1978; 12: 320-8.
10. Cotter LM. The pharmacokinetic of carbamazepine. Eur J Clin Pharmacol 1977; 12:451-6.
11. US Food and Drug Administration. FDA Guidelines, Bioequivalence. US Food and Drug Administration. Division of Bioequivalence. Rockville: Office of Generic Drugs; 1992.
12. FDA (Food and Drugs Administration). US Department of Health and Human Services. Center for Drug Evaluation and Research (CDER). BA and BE studies for orally administered drug products: general considerations. Rockville: Guidance for Industry. Draft Guidance; 1999.
13. Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish interchangeability. Geneva. World Health Organization 2006. Technical Report Series, No 937) Annex. 7.
14. Guidelines for good clinical practice (GCP) for trials on pharmaceutical products. Geneva: World Health Organization; 2006. p. 97-13. (Technical Report Series, No. 850).
15. Jiménez Alemán NM, Calero Carbonell JE, Padrón Yaquis AS. Método analítico por cromatografía líquida de alta resolución para la determinación de carbamazepina en plasma humano. Rev Cubana Farm. 2007 ene.-abr.; 41(1).
16. Steinijans VW, Diletti E. Statistical analysis of bioavailability studies: Parametric and nonparametric confidence intervals. Eur J Clin Pharmacol. 1983;24:127-36.
17. Westlake WJ. Bioavailability and bioequivalence of pharmaceutical formulation. In: Peacee KE. Biopharmaceutical statistics for drug development. New York: Marcel Dekker; 1988. p. 329-52.
18. Shuirmann DJ. A comparison of the two one-side test procedure and the power approach for assessing the equivalence of average bioavailability. J Pharmacoki Biopharm. 1987;15:657-80.
19. Hauschke D, Steinijans VW, Diletti E. A distribution-free procedure for the statistical analysis of bioequivalence studies. Int J Clin Pharmacol Ther Toxicol. 1990;28:72-8.
Recibido: 5 de octubre de 2008.
Aprobado:6 de noviembre de 2008.
Dr. Alejandro Saúl Padrón Yaquis. Centro de Investigación y Desarrollo de Medicamentos (CIDEM). Ave 26, No. 1 605 entre Boyeros y Calzada de Puentes Grandes, CP 10 600, municipio Plaza de la Revolución, La Habana, Cuba. Correo electrónico: cidem@infomed.sld.cu


{kind=link}
{kind=link}