










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Farmacia
versión On-line ISSN 1561-2988
Rev Cubana Farm v.44 n.1 Ciudad de la Habana ene.-mar. 2010
ARTÍCULOS ORIGINALES
Validación del método analítico para el control de la calidad y estudio de estabilidad de propiltiouracilo 50 mg
Analytical method validation for quality control and the study of the 50 mg Propylthiouracil stability
María Olga Valdés BendoyroI; Caridad Margarita García PeñaII; Juan Lugones FernándezIII; Lisandra García BorgesIV; Vivian Martínez EspinosaIV
IMáster en Tecnología y Control de Medicamentos. Empresa Farmacéutica «Reynaldo Gutiérrez». La Habana, Cuba.
IIMáster en Tecnología y Control de Medicamentos. Investigador Agregado. Centro de Investigaciones y Desarrollo de Medicamentos (CIDEM). La Habana, Cuba.
IIIMáster en Tecnología y Control de Medicamentos. Empresa Farmacéutica «Reynaldo Gutiérrez». La Habana, Cuba.
IVTécnico en Tecnología Farmacéutica. CIDEM. La Habana, Cuba.
RESUMEN
Se desarrolló y validó un método analítico por cromatografía líquida de alta resolución, para el control de la calidad y los estudios de estabilidad del propiltiouracilo 50 mg, tabletas. El método se basó en la separación del principio activo a través una columna cromatográfica Lichrospher 100 RP-18 RP-18 (5 µm) (250 x 4 mm), con detección ultravioleta a 272 nm, para lo cual se empleó una fase móvil compuesta por una mezcla desgasificada de solución amortiguadora fosfato de potasio monobásico 0,025 M a pH= 4,6, y acetonitrilo en una proporción de 80:20, con una velocidad de flujo de 0,5 mL/min. El método analítico resultó lineal, preciso, específico y exacto en el intervalo de concentraciones estudiadas.
Palabras clave: Cromatografía líquida de alta resolución, métodos, propiltiouracilo, validación.
ABSTRACT
A high-performance liquid chromatography analytical method was developed and validated for the quality control and stability studies of 50 mg Propylthiouracil tablets. Method is based in active principle separation through a 100 RP-18 RP-18 (5 µm) (250 x 4 mm) Lichrospher chromatography with UV detection to 272 nm, using a mobile phase composed by a ungaseous mixture of a 0.025 M buffer solution-monobasic potassium phosphate to pH= 4,6 ad acetonitrile in a 80:20 ratio with a flux speed of 0,5 mL/min. Analytical method was linear, precise, specific and exact in the study concentrations interval.
Key words: High-performance liquid chromatography, methods, Propylthiouracil, validation
INTRODUCCIÓN
El propiltiouracilo inhibe la síntesis de la hormona tiroidea mediante la inhibición de la incorporación de yodo a la tirosina y del acoplamiento de las iodotirosinas, no interfiere con las acciones de la hormona tiroidea exógena. Es el medicamento de elección en la tormenta tiroidea. Se indica en el tratamiento del hipertiroidismo, e incluye este previo de la cirugía o la radioterapia.1,2
El desarrollo de técnicas o métodos analíticos novedosos o la adecuación de algunos ya reportados es un problema cotidiano. Siempre que esto suceda se exige como parte integral del estudio, la validación del método en cuestión. La validación proporciona un alto grado de confianza y seguridad del proceso productivo o del método analítico así como también en la calidad de los resultados.3,4
Los parámetros analíticos evaluados en la validación del método analítico, fueron exactitud, precisión, especificidad, robustez y linealidad.4-6
El objetivo del presente trabajo fue el desarrollo de un método analítico para la determinación del principio activo en las tabletas de propiltiouracilo 50 mg, por cromatografía líquida de alta resolución (CLAR).
MÉTODOS
La sustancia de referencia química de propiltiouracilo fue suministrada por el grupo de sustancias de referencia del Centro de Investigación y Desarrollo de Medicamentos (CIDEM), La Habana, Cuba, el cual fue analizado por el método cromatográfico establecido para realizar el control de la calidad de la materia prima, con una pureza de 99,6 %. El producto terminado en forma de tabletas, fue elaborado en la Empresa Farmacéutica «Reynaldo Gutiérrez», identificado como el lote P1, fabricado en enero de 2007, con fecha de vencimiento enero de 2009, el cual cumplió con las especificaciones de calidad establecidas para el control de la calidad de las tabletas.
Todos los reactivos utilizados fueron de grado HPLC. En el ensayo se empleó un cromatógrafo (KNAUER) con detector UV/VIS (KNAUER) ajustado a 272 nm, un inyector con un rulo de 20 µL e integrador (SHIMADZU CR 8 A). La separación se realizó isocráticamente empleando una columna Lichrospher 100 RP-18 endcapped (5 µm) (250 x 4 mm), y un flujo de 0,5 mL/min. La fase móvil óptima, consistió en una mezcla del solución amortiguadora fosfato de potasio monobásico 0,025 M a pH= 4,6, y acetonitrilo en una proporción de 80:20.4,5
Para el análisis se disolvió una cantidad exacta de la sustancia de referencia química en agua destilada primeramente y después en fase móvil, de modo que se obtuviera una solución con una concentración final de 50 µg/mL.
Para el análisis de las muestras se seleccionaron aleatoriamente 20 tabletas y se trituraron en mortero hasta obtener un polvo fino. Se pesó exactamente una cantidad del polvo equivalente a 50 miligramos de propiltiouracilo. Se transfirió a un volumétrico de 100 mL. y se adicionaron 10 mL de metanol. Luego se llevó a sonicar por 10 min. Seguidamente se adicionaron 50 mL de agua y se agitó mecánicamente por 20 min. La dilución se realizó con agua y se enrasó y agitó. Luego se procedió a filtrar. Se tomó una alícuota de 10 mL de la solución anterior y se trasvasó a un volumétrico de 100 mL, para diluir con agua hasta el enrase, y se agitó.
Validación del método analítico
La validación fue realizada según la categoría I (USP 30) y la Regulación 41-2007 del Centro para el Control Estatal de la Calidad de los Medicamentos (CECMED), para la validación de métodos de análisis; se evaluaron los parámetros que a continuación se describen:3-5
Linealidad
Para el análisis de la linealidad se realizó el modelo de 3 determinaciones para 5 concentraciones diferentes: 60, 80, 100,120, y 140 %. Se determinó la ecuación de la recta, el coeficiente de correlación, la prueba de significación estadística de significación de la pendiente Sb rel(%), los coeficiente de variación de los factores de respuesta, ensayo de proporcionalidad.
Exactitud
Para el análisis de exactitud se realizó el modelo de 3 réplicas para 3 concentraciones diferentes: 80, 100, 120 %, se determinó el % de recuperación, la desviación estándar y el coeficiente de variación. Se aplicó además el ensayo de Gochran con vistas a comprobar si la variación de la concentración producía diferencias significativas en los resultados y la prueba t de Student para determinar diferencias significativas entre la recuperación media y el 100 %.
Precisión
Para el estudio de la precisión se aplicó el modelo de repetibilidad con 6 réplicas. Con ellas se determinaron los valores medios, la desviación estándar y el coeficiente de variación.
De igual manera para el ensayo de la precisión intermedia se utilizaron 3 valores de concentración que correspondieron al 80, 100 y 120 % para 2 analistas y 3 días diferentes. Se aplicó la prueba de Fisher y t de Student para determinar si existían diferencias significativas entre los resultados al variar las condiciones de análisis.
Especificidad
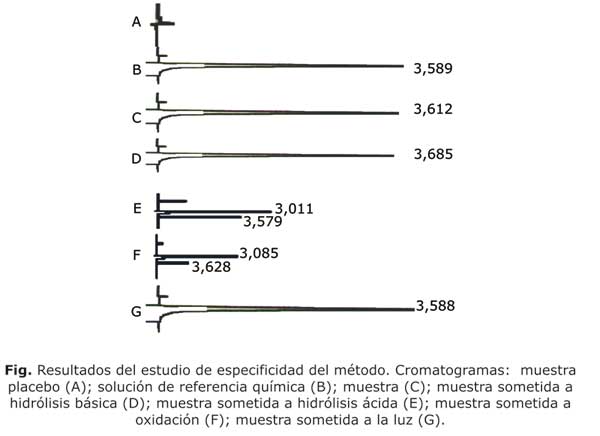
Para el estudio de especificidad se analizaron: la sustancia de referencia del propiltiouracilo, el placebo; las muestras de producto terminado y muestras sometidas a condiciones drásticas como: hidrólisis ácida HCl 3 N, hidrólisis básica NaOH 3 N, oxidación H2O2 y a la luz durante 3 días.
Criterio de aceptación: No se debían obtener señales del placebo y de los productos de degradación en la zona de elusión del principio activo. Las áreas bajo las curvas del patrón y del propiltiouracilo en el producto terminado debían ser similares.
RESULTADOS
La figura muestra los resultados del estudio de especificidad del método. Como se observa en el cromatograma correspondiente a la muestra placebo (A) no se obtuvo ninguna señal en la zona de interés, al ser comparado con la señal obtenida para la solución estándar de referencia (B) y de la muestra de propiltiouracilo tabletas (C), lo cual indica que los excipientes o sustancias auxiliares de la solución no interfieren en la determinación del principio activo. En cuanto a las muestras sometidas a condiciones drásticas de: hidrólisis básica y hidrólisis ácida, cromatogramas (D, E), la molécula resultó ser bastante estable, solo se pudo apreciar una ligera degradación por la disminución de la altura de los picos y la aparición de pequeños picos secundarios como posibles productos de degradación, el cual no interfiere en la determinación del principio activo.
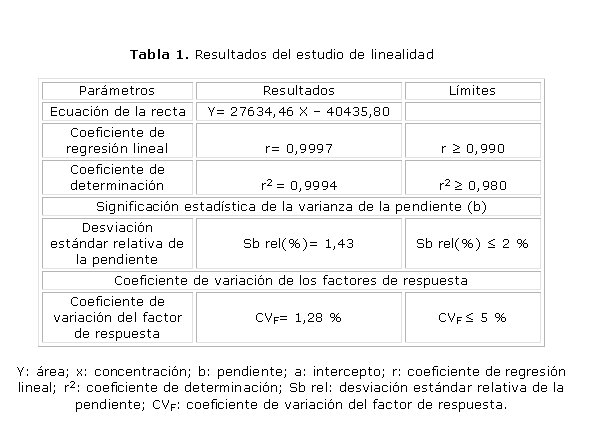
En la tabla 1 se reportan los resultados de los estudios de la linealidad del sistema; el coeficiente de regresión lineal fue de 0,9997 y el coeficiente de variación del factor de respuesta resultó igual a 1,28 %.
Los resultados del estudio de precisión del método aparecen reportado en la tabla 2. En el estudio de repetibilidad realizado, la media obtenida fue de 98,15 % y el coeficiente de variación fue de 0,79 %, mientras que los valores de F calculadas y los valores de t calculadas resultaron menores que los valores tabulados, para un 95 % de confianza, para cada uno de los niveles estudiados.
En la tabla 3 aparece reportado los resultados del estudio de exactitud. La recuperación media fue de 100,08 % y el valor de t calculada (1,411) y de G calculada (0,440) fueron menores que los valores tabulados, para un 95 % de confianza, t tabulada 2,306 y G tabulada 0,797.
DISCUSIÓN
Los resultados del estudio de especificidad del método (figura), demuestran la especificidad del método al no presentarse interferencias de picos adicionales en la zona de elución del producto principal, ya que los productos de degradación y los excipientes de la formulación presentan tiempos de retenciones diferentes al principio activo, todos inferiores al tiempo de retención del propiltiouracilo. Este estudio permite concluir que el método cromatográfico resultó ser específico pues permite cuantificar el principio activo después de la degradación de este en muestras de medicamento sin interferencias de excipientes y/o producto de degradación.4
Los resultados del estudio de linealidad muestran coeficientes de regresión y de determinación superiores a los exigidos, 0,99 y 0,98 respectivamente; se demostró con el valor del coeficiente de correlación obtenido, cercano a la unidad, la existencia de correlación con una probabilidad elevada, así como el grado de relación entre las variables concentración y respuesta detectada por el equipo empleado. También el coeficiente de variación de los factores de respuesta y la desviación estándar relativa de la pendiente fueron inferiores al normado como máximo para estos indicadores: 5 y 2 %, respectivamente, ambos considerados estimadores puntuales que permiten caracterizar la variabilidad. El valor obtenido del CVf permitió demostrar que existe variabilidad en la relación respuesta y concentración para cada nivel evaluado. El intervalo de confianza del intercepto incluye al cero, lo que permite excluir la significación del error del intercepto. Se demostró con estos resultados la linealidad del método propuesto.
En el estudio de la repetibilidad (tabla 2) realizado a una misma muestra, por el mismo analista, el mismo día, a través de seis réplicas, se alcanzó un coeficiente de variación adecuado (0,79 %), lo que demostró la buena precisión del método; se observó una variabilidad de los resultados dentro de los límites establecidos para los métodos cromatográficos: CV £ 2,0 %.6
Los valores que se obtienen en el estudio de precisión intermedia, de las pruebas de Fischer y de la t de Student, para el estudio de la precisión intermedia demostraron que no existían diferencias significativas entre las precisiones alcanzadas por los analistas en diferentes días para una probabilidad de 0,05 %, ya que el valor de Fcalculada es menor que la Ftabulada, este resultado permite establecer que las precisiones son similares (tabla 2). Al realizar la prueba t de Student el valor calculado resultó menor que el tabulado, para una probabilidad de 0,05, lo cual demostró que no existían diferencias significativas entre las medias alcanzadas, con un nivel de significación de un 5 %.
Los valores de porcentaje de recobrado estuvieron dentro de los límites establecidos para los métodos cromatográficos (98-102 %) y los valores del coeficiente de variación para cada uno de los valores de concentración estudiados resultaron ser menor que el 2 %.4 En la influencia del factor concentración sobre la variabilidad de los resultados de la exactitud al aplicar la prueba de Cochran, se obtuvo que G calculada fue menor que G tabulada para una probabilidad de 0,05, k= 3 y n= 3; por lo tanto, las varianzas de las concentraciones empleadas son equivalentes, lo cual indica que la concentración no influye en la variabilidad de estos. Al realizar la prueba de significación entre la recuperación media y el 100,0 % de recuperación, con un coeficiente de variación de 0,98 %, se obtuvo una t calculada menor que t tabulada.6 Los resultados demostraron la capacidad del método para dar resultados cercanos al valor verdadero, observándose una buena exactitud.
El método analítico validado, por CLAR, para la cuantificación del principio activo de las tabletas de propiltiouracilo 50 mg para el control de la calidad y el estudio de estabilidad resultó ser lineal, preciso, exacto, robusto y específico, en el intervalo de concentraciones establecido del 60 al 140 %.
REFERENCIAS BIBLIOGRÁFICAS
1. Goodman A, Gilman A. Las bases farmacológicas de la terapéutica. Tomo II. 3ra ed. La Habana: Editorial Científico Técnica; 1994. p. 466-7. (Edición Revolucionaria).
2. PDR. Physician´s Desk Reference. 57 ed. New York: Inc at Montuale; 2003. p. 332, 2193, 2905, 3270.
3. United States Pharmacopoeial Convention. USP XXX. United States Pharmacopoeia Convention. 30 ed. Rockville: Mack Printing; 2007. p. 2883-5. Versión electrónica en CD.
4. Quattrocchi OA, Laba RF. Introducción a la HPLC en Aplicación y práctica. Buenos Aires: Ed. Artes Gráficas Farro; 1992. p. 106-22, 284, 302-28.
5. Dierksneier G. Métodos cromatográficos. La Habana: Ed. Científico Técnica; 2005. p. 1-4, 256-412.
6. Validation of Analytical Procedures. Technical Requirements for the Registration of Pharmaceuticals For Human Use. International Conference on Harmonization, ICH-Q2A, Geneva. 1995
Recibido: 18 de septiembre de 2009.
Aprobado: 21 de octubre de 2009.
M. C. Caridad Margarita García Peña. Centro de Investigación y Desarrollo de Medicamentos (CIDEM). Ave 26, No. 1 605 entre Boyeros y Calzada de Puentes Grandes, CP 10 600, municipio Plaza de la Revolución, La Habana, Cuba. Correo electrónico: caridadgp@infomed.sld.cu


{kind=link}
{kind=link}
{kind=link}
{kind=link}