










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Farmacia
versión On-line ISSN 1561-2988
Rev Cubana Farm v.44 n.2 Ciudad de la Habana abr.-jun. 2010
ARTÍCULOS DE REVISIÓN
Nuevos enfoques sobre la función de las lipoproteínas plasmáticas en las enfermedades de origen aterotrombótico
New approaches on the function of plasmatic lipoproteins in diseases of atherothombosis origin
Livan Delgado-RocheI; Gregorio Martínez-SánchezII
ILicenciado en Ciencias Farmacéuticas. Centro de Estudios para las Investigaciones y Evaluaciones Biológicas. Instituto de Farmacia y Alimentos. Universidad de La Habana. La Habana, Cuba.
RESUMEN
La aterosclerosis y las complicaciones que de ella se derivan representan una de las causas más frecuentes de muerte en el mundo occidental. Esta es una enfermedad crónica y progresiva, la cual responde a daños multifactoriales a la pared de los vasos sanguíneos. Estos daños promueven la formación de placas ateromatosas y fibróticas, las cuales constituyen regiones engrosadas de la capa íntima de las arterias y están formadas por tejido fibrótico, células inmunocompetentes y del plasma, así como por lípidos. El colesterol de lipoproteínas de baja densidad y la modificación oxidativa que estas partículas sufren, constituyen el centro de las hipótesis formuladas en torno a la aterogénesis. En tanto, su contraparte biológica, las lipoproteínas de alta densidad han sido catalogadas como antiaterogénicas y se ha asumido que su disminución constituye un factor de riesgo cardiovascular; sin embargo, recientemente se ha podido comprobar que estos criterios no son absolutos. Ensayos clínicos realizados han demostrado que los efectos beneficiosos del colesterol de lipoproteínas de alta densidad dependen principalmente de su calidad y no de su cantidad. En el presente trabajo se presentan nuevos fundamentos acerca la función de estas moléculas en la aterosclerosis.
Palabras clave: Aterosclerosis, factores de riesgo cardiovascular, lipoproteínas de alta densidad, lipoproteínas de baja densidad, estrés oxidativo, inflamación.
ABSTRACT
Atherosclerosis and its inherent complications are one of the more frequent death causes in western world because it is a chronic and progressive disease, which provokes many damages to blood vessels wall. These damages promote the formation of atheromatous and fibrotic plaques, which are thickened regions of the intima of arteries and are formed by fibrotic tissue, plasma and immunocompetent cells, as well as by lipids. Cholesterol of low-density lipoproteins and the oxidative modification of these particles are the center of the hypotheses formulated around the atherogenesis. While, its biological counterpart, the high-density lipoproteins have been classified as antiatherogenous and has been assumed that its decrease is a cardiovascular risk factor, however, recently, has been possible to prove that these criteria aren't absolutes. The clinical trials performed have demonstrated that beneficial effects of cholesterol of high-density lipoproteins depend mainly on its quality and not on its quantity. In present paper are showed new reasons on the function of theses molecules in atherosclerosis.
Key words: Atherosclerosis, cardiovascular risk factors, high-density lipoproteins, low-density lipoproteins, oxidative stress, inflammation.
INTRODUCCIÓN
La aterosclerosis y las complicaciones que de ella se derivan representan una de las causas más frecuentes de muerte en el mundo occidental .1 Esta es una enfermedad vascular crónica en la cual la inflamación y el estrés oxidativo (EO) desempeñan una función importante en cada una de sus etapas. El desarrollo y progreso de los procesos aterogénicos constituyen un reflejo de la deposición de colesterol en la capa íntima de las grandes arterias. Este depósito de colesterol promueve la fase de activación endotelial, inducida por citocinas pro-inflamatorias, lipoproteínas de baja densidad oxidadas (LDL-ox) y cambios en el flujo hemodinámica.2 Estos factores promueven la expresión de moléculas de adhesión endotelial y quimiocinas, seguido del reclutamiento y activación de monocitos y linfocitos circulantes. La disfunción endotelial, uno de los eventos tempranos que tienen lugar en la aterosclerosis, afecta el delicado equilibrio que debe existir entre factores vasodilatadores y vasoconstrictores.3 Las manifestaciones clínicas de esta enfermedad, entre las que se encuentran los síndromes coronarios agudos, son la expresión de la erosión y ruptura de la placa ateroesclerótica, formándose trombos que provocan la oclusión de la luz arterial.4
En la actualidad, los postulados de mayor aceptación a la hora de dar explicación a los fenómenos que están implicados en la aterosclerosis se centran en el proceso de oxidación de las lipoproteínas de baja densidad (LDL).5 Las LDL-ox estimulan el reclutamiento de monocitos circulantes y su diferenciación en macrófagos residentes, además de poseer propiedades citotóxicas para el endotelio y otras células vasculares.6
Existen otros tipos de lipoproteínas que pueden incidir sobre el proceso de aterogénesis y tener un comportamiento antiaterogénico o proaterogénico. Su clasificación se realiza atendiendo a su densidad, composición lipídica y tipo de apoproteína que las componen. De este modo se pueden identificar de forma general quilomicrones, lipoproteínas de muy baja densidad (VLDL), LDL, lipoproteínas de densidad intermedia (IDL) y lipoproteínas de alta densidad (HDL).7 Sus efectos antes mencionados se abordarán más adelante en este trabajo.
ASPECTOS FISIOPATOLÓGICOS DE LA ATEROSCLEROSIS
La aterosclerosis es una enfermedad crónica y progresiva la cual responde a daños multifactoriales a la pared de los vasos sanguíneos. Estos daños promueven la formación de placas ateromatosas y fibróticas, las cuales constituyen regiones engrosadas de la capa íntima de las arterias y están formadas por tejido fibrótico, células inmunocompetentes y del plasma, así como por lípidos. El daño a la pared arterial provoca disfunción endotelial y el incremento de la adhesión leucocitaria y de plaquetas al endotelio vascular. Durante este proceso se liberan una serie de mediadores inflamatorios que potencian la proliferación de las células del músculo liso vascular (MLV), la acumulación de lípidos peroxidados y la subsecuente formación de la placa de ateroma.8
El endotelio vascular constituye una monocapa de células que recubre el lumen de los vasos sanguíneos. Las células endoteliales desempeñan una función muy importante desde le punto de vista estructural pero también funcional. Estas contribuyen al mantenimiento del tono vascular, regulan la homeodinámica intravascular y la permeabilidad, protegen contra el EO e inhiben la adhesión y migración celular.9 Los avances en la biología vascular han permitido identificar una serie de sustancias vasoactivas liberadas por el endotelio. Estas sustancias pueden clasificarse en 2 grupos: los factores relajantes derivados del endotelio (EDRF) y los factores constrictores derivados del endotelio (EDCF). Se ha demostrado que los EDRF como el óxido nítrico (NO.) y el factor hiperpolarizante derivado del endotelio (peróxido de hidrógeno) protegen la vasculatura contra el daño aterogénico, mientras que los EDCF se oponen a este efecto y promueven la progresión de enfermedades cardiovasculares (ECV).8 En la disfunción endotelial se produce un desequilibrio entre estos factores, caracterizado por una disminución en la biodisponibilidad de NO. fundamentalmente y un incremento de los EDCF.10 Este desequilibrio provoca una afectación en la vasodilatación dependiente del endotelio, lo cual representa la manifestación funcional más importante en la disfunción endotelial. Además, se puede producir un estado de activación endotelial, caracterizado por la proliferación celular, la inflamación y fenómenos pro-coagulantes.11
La inflamación vascular desempeña una función fundamental durante las diferentes etapas del proceso aterogénico, desde la iniciación hasta la progresión y ruptura de la placa, responsable esta última de los síndromes coronarios agudos.12-14 La acumulación de LDL-ox promueve la migración de monocitos y linfocitos T, así como la producción por parte de las células endoteliales de moléculas de adhesión como ICAM-1, VCAM-1, P-selectina o E-selectina; de sustancias quimiotácticas como la proteína quimiotáctica de macrófagos 1 (MPC-1) y factores de crecimiento como el factor estimulante de colonias. Por su parte, los macrófagos activados expresan en su membrana receptores scavenger, los cuales facilitan la internalización no regulada de colesterol y la subsecuente formación de células espumosas 8. Adicionalmente, los macrófagos producen metaloproteinasas que degradan la matriz celular y contribuyen a la ruptura de la placa. Una vez ocurrido esto se produce una activación de la cascada de la coagulación por parte de proteínas titulares, así como de plaquetas, leucocitos, neutrófilos y otras células que propician la formación del trombo.15-17
Entre los marcadores de la respuesta inflamatoria que reflejan el proceso aterogénico se pueden citar citocinas pro-inflamatorias como la IL-6 y el factor de necrosis tumoral alfa (TNF-a
), moléculas de adhesión y proteínas de fase aguda como la proteína C reactiva (PCR) y el amiloide sérico A.18El EO constituye otro de los fenómenos que están involucrados en la patogénesis de la aterosclerosis. Un número creciente de estudios han postulado la participación del EO en el desarrollo del proceso aterogénico, sobre todo en la disfunción endotelial.19 Los organismos aerobios son capaces de producir especies reactivas del oxígeno (ERO) a partir de la reducción incompleta del oxígeno molecular. Estas se pueden formar mediante reacciones enzimáticas de la NADPH oxidasa, xantina oxidasa, lipoxigenasa y ciclooxigenasa, entre otras. Otras fuentes generadoras de ERO son los procesos de auto-oxidación de diversos sustratos y la cadena de transporte electrónico mitocondrial, esta última constituye la vía fundamental de generación intracelular de ERO y es capaz de mediar procesos de EO y muerte celular.1,20
A partir de los procesos antes mencionados se forma el radical anión superóxido (O2·-) el cual es reducido a peróxido de hidrógeno (H2O2) a través de la acción de la enzima superóxido dismutasa (SOD) o de forma espontánea. A su vez el H2O2 puede descomponerse a radical hidroxilo (·OH) mediante la acción de metales de transición como el hierro y el cobre. Esta ERO produce daños considerables sobre lípidos, proteínas y ácidos nucleicos.21,22 El efecto perjudicial de estas especies químicas de alta relatividad e inestabilidad sobre la función vascular puede responder a varios mecanismos. Primeramente, pueden oxidar biomoléculas, pueden interactuar con mediadores vasoactivos endógenos formados por las células endoteliales como el NO· y además son capaces de inducir procesos de peroxidación lipídica, lo cual favorece la formación de LDL-ox, uno de los mediadores más importantes de la aterosclerosis.19 Mientras que la LDL nativa no es capaz de provocar acumulación de ésteres del colesterol en macrófagos, la LDL-ox sí y adicionalmente interviene en otros mecanismos presentes en el desarrollo de la aterogénesis como es la citotoxicidad y las acciones quimiotácticas sobre monolitos.23,24 De esta manera se puede plantear que el EO constituye un aspecto de gran importancia a la hora de trazar estrategias de intervención nutricional y/o farmacológica para el tratamiento de esta enfermedad.
Como se ha mencionado anteriormente, el proceso ateroesclerótico no solo afecta al endotelio vascular sino que también compromete el adecuado funcionamiento de las células del MLV.25 La disfunción de estas provoca alteraciones en la contractilidad y promueve procesos proliferativos. En el 2006, Inoue y Node plantearon la necesidad de no ver como entidades independientes los procesos de disfunción del endotelio y del MLV, por ello propusieron un nuevo concepto: la insuficiencia vascular, a la cual definieron como la integración entre estos dos procesos y los cambios metabólicos que tienen lugar.8 En la figura se puede observar de forma esquemática el planteamiento realizado por los autores.
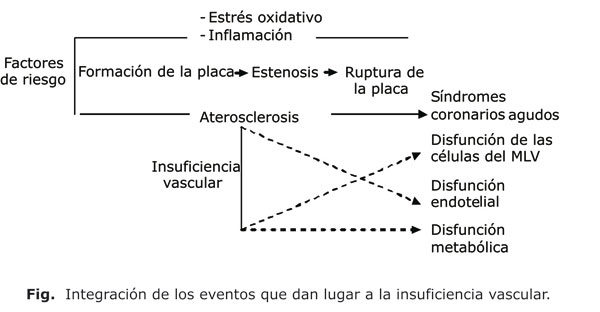
La insuficiencia vascular comprende desde los procesos de iniciación de la aterosclerosis en pacientes con factores de riesgo aterotrombóticos hasta el establecimiento y progreso de la enfermedad. El status fisiopatológico de la insuficiencia vascular enmarca desde los estados subclínicos de la enfermedad con afectaciones de la función endotelial y la relajación del MLV hasta estados avanzados de la enfermedad con procesos de proliferación celular, fibrosis y formación de la placa de ateroma.8 Para poder llevar a cabo intervenciones farmacológicas es necesario estudiar y caracterizar cada una de las etapas de la insuficiencia vascular; para ello sería indispensable medir la función endotelial y de las células del MLV, el perfil lipídico, marcadores del proceso inflamatorio y de EO. Todo cuanto se realice en este sentido impactará de forma positiva sobre la disminución de la morbilidad y mortalidad debida a síndromes coronarios agudos y otros eventos cardiovasculares adversos.
LIPOPROTEÍNAS DE BAJA DENSIDAD Y ENFERMEDADES ATEROTROMBÓTICAS
El papel del colesterol de LDL (c-LDL) en la patogénesis de las ECV, así como el efecto beneficioso de su reducción en pacientes con riesgo cardiovascular ha sido bien establecido.7 La entrada de colesterol a la célula se produce a través de un mecanismo perfectamente regulado dependiente de su contenido intracelular. Este es capaz de regular la actividad del receptor de LDL (r-LDL) y de las enzimas involucradas en el metabolismo del colesterol como son la 3-hidroxi-3-metilglutaril coenzima A reductasa (HMG-CoA reductasa), colesterol aciltransferasa (ACAT) y la colesteril éster hidrolasa neutra (NCEH).26 Sin embargo, la modificación oxidativa de las LDL provoca que estas no sean reconocidas por el r-LDL sino por una familia de receptores de membrana diferentes a este, denominados receptores scavenger. Estos están localizados fundamentalmente en macrófagos y no son regulados por el contenido de colesterol intracelular. En esta familia de al menos 11 miembros, fundamentalmente el SR-A y el CD 36 son los responsables de la captación endocítica de las LDL-ox.27 Todo ello resulta en una acumulación anómala de colesterol y en la formación de células espumosas, típicas de las estrías grasas que pueden ser observadas en las lesiones ateroscleróticas.26
Durante los procesos de oxidación del colesterol se forman una serie de metabolitos conocidos como oxisteroles.28 Estos productos de oxidación, especialmente el 7b
-hidroxicolesterol (7b-OH) y 7-cetocolesterol (7-ceto) son los componentes tóxicos mayoritarios encontrados en las LDL-ox.29 Estudios recientes han revelado que ambos oxisteroles son capaces de inducir procesos de muerte celular mediados por necrosis y apoptosis. Estos efectos están en estrecha relación con incrementos en la generación de ERO, depleción de grupos tioles, así como la permeabilización de las membrana lisosomal y mitocondrial.30 Estudios in vitro sugieren que la combinación de estos oxisteroles presenta un sinergismo citotóxico, lo cual apoya la hipótesis de que el riesgo aterogénico depende en gran medida de la relación entre estos 2 componentes de la LDL-ox.31 La profundización en el estudio de los efectos biológicos de los diferentes tipos de oxisteroles identificados, puede constituir un avance promisorio en cuanto a las estrategias terapéuticas que se deben seguir en esta y otras enfermedades degenerativas en las cuales están involucradas.Estudios arteriográficos realizados en diferentes ensayos clínicos han demostrado que la disminución del c-LDL reduce la progresión de las lesiones ateromatosas. Sin embargo, en un número considerable de pacientes, a pesar de disminuir las concentraciones de c-LDL, ha persistido la progresión de la enfermedad.32 Estas observaciones pueden estar relacionadas con la heterogeneidad de las LDL plasmáticas en humanos. Estas incluyen diferentes subclases, las cuales pueden identificarse mediante ultracentrifugación o por el gradiente electroforético en gel de poliacrilamida.33 Estos tipos de métodos han permitido distinguir 7 subclases de acuerdo con su diámetro: LDL-I (27,2-28,5 nm), LDL-IIa (26,5-27,2 nm), LDL-IIb (25,6-26,5 nm), LDL-IIIa (24,7-25,6 nm), LDL-IIIb (24,2-24,7 nm), LDL-IVa (23,3-24,2 nm) y LDL-IVb (22,0-23,3 nm).34,35 Por otra parte, desde el punto de vista fenotípico se han clasificado como LDL patrón A (partículas grandes con un diámetro de 262 Å o mayor) y LDL patrón B (partículas pequeñas con un diámetro de 257 Å o menor).7
Aunque históricamente se ha considerado el aumento en la concentración de c-LDL como un factor de riesgo cardiovascular, la talla de las LDL parece emerger como un nuevo e importante marcador de riesgo para estas enfermedades.36 Evidencias científicas sugieren que las LDL pequeñas (fenotipo B) así como el número de estas pueden incrementar la susceptibilidad de desarrollo de lesiones ateromatosas.37 En modelos de la enfermedad en conejos se ha podido observar que esta subclase pequeña y densa de LDL penetra a la pared arterial de aortas lesionadas 50 % más rápido que otros tipos de LDL. Igual comportamiento se apreció en zonas no lesionadas de aortas, en este caso la internalización de las LDL pequeñas densas fue un 90 % más rápida comparada con el patrón A (LDL grande) (p < 0,01).38 Por otra parte, en pacientes que presentan el patrón LDL B se produce una síntesis mayor de tromboxanos, una disminución de ácido siálico y un aumento del enlazamiento a proteoglicanos.39 Por último, otras evidencias reflejan que en estos pacientes las concentraciones de vitamina E son bajas, la susceptibilidad al EO y una lipemia pospandrial mayor comparada con pacientes del fenotipo A.40
La medición del número y talla de las partículas de LDL emerge como una herramienta potencial para predecir el riesgo de padecer un evento cardiovascular adverso, así como para la selección de una terapia individualizada según las características del paciente.
LOPOPROTEÍNAS DE BAJA DENSIDAD OXIDADAS Y FACTORES DE DE TRANSCRIPCIÓN PRO-INFLAMATORIOS
Entre los efectos atribuidos a las LDL-ox se puede citar la estimulación de la producción de citocinas proinflamatorias y factores quimiotácticos.41,42 Muchos de estos efectos están mediados por rutas de señalización celular, especialmente a través de la activación de factores de transcripción, que a su vez son capaces de promover la expresión de genes relacionados con la respuesta inflamatoria y el EO.42
Uno de los factores de transcripción proinflamatorios que son activados por la LDL-ox es el activador de proteínas 1 (AP1).43 Este está constituido por c-fos/c-jun y está implicado en el control del crecimiento y la carcinogénesis. El AP1 también regula la expresión de genes que codifican para citocinas proinflamatorias como el factor de necrosis tumoral alfa (TNF-a
),43,44 interleucina 1 beta (IL1b)45 y osteopontina.46 También cabe destacar que la actividad de AP1 está bajo el control del estado redox celular y se conoce que es estimulada por ERO como el radical ·OH.42La activación de AP1 por la LDL-ox ha sido demostrada en células del MLV,47 fibroblastos y células endoteliales.48 En la aterosclerosis este factor de transcripción ha sido relacionado con los procesos de proliferación y reestenosis.49 También se ha demostrado que AP1 activada induce procesos inflamatorios mediados por angiotensina II en ratas50 y modula los niveles intracelulares de colesterol a través de la expresión de ABCA1, un transportador de membrana que facilita el flujo de colesterol a través de esta .51
En tanto, otro de los mediadores que son activados por la LDL-ox es el factor de transcripción nuclear k
B (NFkB), lo cual fue reportado por primera vez por Rajavashisth y otros.52 En macrófagos RAW 264.7 fue reportado que la LDL-ox y su constituyente lisofosfatidilcolina activan NFkB a través de una proteína cinasa C y/o rutas calcio dependiente y que este fenómeno no involucra el procesamiento endocítico de la LDL-ox.53 Este factor de transcripción nuclear está involucrado en varias etapas del progreso de la aterosclerosis, entre los las que se pueden citar la adhesión de monocitos, formación de células espumosas y la inflamación.54 Estos efectos del NFêB han sido demostrados mediante su identificación en la forma activa en placas ateromatosas y por el hecho de que la inhibición selectiva de este tributa en una reducción de la formación de células espumosas.55,56 También se ha observado que las estatinas son capaces de disminuir la activación del NFkB en células endoteliales.57 Existen otros factores de transcripción proinflamatorios que también son activados por la LDL-ox como es el caso de STAT 1/3, NFAT, HIF1a, Sp1, entre otros.41
LIPOPROTEÍNAS DE ALTA DENSIDAD: NUEVOS ENFOQUES SOBRE SU FUNCIÓN EN LA ATEROGÉNESIS
Históricamente se ha considerado a las HDL como el colesterol «bueno», sobre todo por su capacidad de transportarlo desde los tejidos periféricos hacia el hígado para su catabolismo. La apoproteína A1 (ApoA1) constituye el 70 % de las HDL lo cual le confiere propiedades antioxidantes que previenen la oxidación de las LDL, así como propiedades antiinflamatorias entre las que se incluye la inhibición de la expresión de moléculas de adhesión endotelial.58,59 En el hígado se produce la síntesis y secreción de ApoA1, partícula que es conocida como pre-b
HDL y que interviene en el transporte reverso del colesterol, proceso mediante el cual el colesterol es transportado desde los tejidos periféricos hacia el hígado para su catabolismo y excreción. Esta partícula pobre en su contenido lipídico es convertida por acción de la lecitina colesterol aciltransferasa (LCAT) a su forma madura, lo que incrementa su capacidad de almacenamiento.60Las propiedades antioxidantes de la HDL son atribuidas tanto a la ApoA1 como a la enzima paraoxonasa (PON) y al factor activador de plaquetas acetil hidrolasa (PAF-AH).61 Estos actúan como fosfolipasas e hidrolizan los fosfolípidos oxidados y de esta manera reducen el número de estos productos de oxidación contenidos en las LDL y VLDL.62 En virtud a la capacidad de las HDL de prevenir los procesos de peroxidación lipídica (POL), reducen la aterogenicidad de la apoproteína B (ApoB) contenida en las LDL. Adicionalmente, reducen los procesos inflamatorios inducidos por las LDL-ox e inhiben de manera efectiva la unión de monocitos circulantes a moléculas de adhesión endotelial.60
Sin embargo, en los últimos años ha sido aceptado el criterio de que las HDL pueden volverse no funcionales bajo algunos estados de enfermedades como es el caso de las cardiovasculares.63 El proceso inflamatorio que tiene lugar durante la aterogénesis influye sobre la función de estas lipoproteínas. La inducción de una respuesta de fase aguda provoca cambios estructurales en las HDL; fundamentalmente en proteínas asociadas a estas. Uno de estos cambios lo constituye la pérdida de ApoA1 y la PON y en su lugar se incorporan proteínas reactivas de fase aguda como la PCR, el amiloide A sérico y otras.64 La incorporación de estas proteínas de fase aguda a las partículas de HDL, se piensa que provoca la pérdida de algunas funciones como pueden ser: 1) transporte reverso de colesterol, 2) inhibición de la oxidación de las LDL, 3) supresión de la respuesta inflamatoria frente a las LDL-ox.65 Estas funciones de las HDL también pueden ser modificadas por acción de la mieloperoxidasa (MPO), esta enzima presente en macrófagos ha sido identificada como responsable de la modificación oxidativa de ApoA 1 a través de la formación de ácido hipocloroso (HOCl-).66 Esta oxidación resulta en una afectación del transporte reverso del colesterol por parte de las HDL, de ahí que la actividad aumentada de esta enzima se ha considerado como un factor de riesgo cardiovascular (FRCV).67
Estudios de Framingham mostraron que el 40 % de los accidentes cardiovasculares ocurridos en un grupo de pacientes cardiópatas tenían valores normales e incluso elevados de HDL, lo cual sugiere que las propiedades funcionales de las HDL, más que su concentración total en sangre, son las responsables de sus propiedades antiaterogénicas.68 Por tales motivos se han desarrollado y estandarizado métodos para evaluar la calidad funcional de HDL. Van Lenten y colaboradores diseñaron un método basado en la capacidad de las HDL de inhibir la actividad quimiotáctica de monocitos en cultivo de células endoteliales. Por otra parte, se ha visto que en modelos experimentales de aterosclerosis en conejos hay un aumento de marcadores de inflamación asociado a una función anormal de HDL; y estos índices proinflamatorios mostraron correlación positiva con las concentraciones de la proteína de fase aguda amiloide A sérico. A partir de estos nuevos conocimientos se pone a disposición de los profesionales que trabajan en este campo de investigación, herramientas útiles que les permitirá realizar estudios en pos de mejorar la calidad de vida de los pacientes que se encuentran afectados por estas enfermedades.
El colesterol de HDL (HDL-c) parece ser una diana terapéutica muy activa y plausible en el tratamiento de las ECV, sin embargo, resulta de vital importancia comprender que no todas las partículas de esta familia son iguales en estructura y función. Por ejemplo, la misión de la HDL-c naciente (ApoA-1 pobre en lípidos, fase inicial de la HDL-c) es retirar colesterol de los tejidos extrahepáticos y devolverlo al hígado, mientras que las HDL-c en su etapa final (HDL-c esférico y lleno de lípidos) todavía no han sido bien comprendidos. También es clave el hecho de que el incremento de HDL-c por diferentes mecanismos no tiene por qué llevar a los mismos efectos. El reciente fracaso de un ensayo clínico con un inhibidor de CETP torcetrapib no debe ser considerado como el fracaso de todas las terapias que incrementan las HDL-c en función de reducir los riesgos de ECV, sino que debe ser tomada más bien como una prueba de que los efectos beneficiosos del HDL-c dependerán principalmente de su calidad y no de su cantidad.68
REFERENCIAS BIBLIOGRÁFICAS
1. Paim BA, Velho JA, Castillo RF, Oliveira HCF, Vercesi AE. Oxidative stress in hypercholesterolemic LDL (low-density lipoprotein) receptor knockout mice is associated with low content of mitochondrial NADP-linked substrates and is partially reversed by citrate replacement. Free Radic Biol Med. 2008;44:444-51.
2. Dogné JM, Hanson J, Pratico D. Thromboxane, prostacyclin and isoprostanes: therapeutic targets in atherogenesis. TRENDS Pharmacol Sci. 2005;26(12):639-44.
3. Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. New Engl J Med. 2005;352:1685-95.
4. Steinberg D. Atherogenesis in perspective: hypercholesterolemia and inflammation as partners in crime. Nat Med. 2002;8:1211-27.
5. Chisolm GM, Steinberg D. The oxidative modification hypothesis of atherosclerosis: an overview. Free Radic Biol Med. 2000;28:1815-26.
6. Libby P. Atherosclerosis: disease biology affecting the coronary vasculature. Am J Cardiol. 2006;98:3Q-9.
7. Superko HR, Gadesam RR. Is it LDL particle size or number that correlates with risk for cardiovascular disease? Cur Atheroscler Rep 2008; 10:377-385.
8. Inoue T, Node K. Vascular failure: a new clinical entity for vascular disease. J Hypertens. 2006;24:2121-30.
9. Luscher TF, Barton M. Biology of the endothelium. Clin Cardiol. 1997;20:3-10
10. Lerman A, Burnet JC Jr. Intact and altered endothelium in regulation of vasomotion. Circulation. 1992; 86(Suppl III):12-9.
11. Anderson TJ. Assessment and treatment of endothelial dysfunction in humans. J Am Coll Cardiol. 1999; 34:631-8.
12. Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135-43.
13. Lusis AJ. Atherosclerosis. Nature. 2004;407:233-41.
14. Shishehbor U, Sukhova GK, Shimizu K, Mach F, Libby P. Inhibition of CD40 siganling limits evolution of stablished atherosclerosis in mice. Proc Natl Acad Sci USA. 2000;97:7458-63.
15. Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the unstable plaque. Prog Cardiovasc Dis. 2002;44:349-56.
16. Ott I, Newmann FJ, Gawaz M, Schmitt M, Schomig A. Increased neutrophil-platelet adhesion in patients with unstable angina. Circulation. 1996; 94:1230-46.
17. Furman MI, Benoit SE, Barnard MR, Valeri CR, Borbone ML, Becker RC, et al. Increased platelet reactivity and circulating monocyte-platelet aggregates in patients with stable coronary artery disease. J Am Coll Cardiol. 1998; 31:352-8.
18. Ridker PM, Stampfer MJ, Rifai N. Novel risk factors for systemic atherosclerosis: a comparison of C-reactive protein, fibrinogen, homocysteine, lipoprotein (a), and standard cholesterol screening as predictors of peripheral arterial disease. JAMA. 2001;285:2481-5.
19. Steinberg D. Low density lipoprotein oxidation and its pathological significance. J Biol Chem. 1997;272:20963-6.
20. St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem. 2002;277:44784-90.
21. Martínez G, Popov I, Pérez G, Al-Dalain SM, Horwat R, Giuliani A, et al. Contribution to characterization of oxidative stress in diabetic patients with microangiopatic complications. Acta Farm Bonaerense. 2005;24(2):197-203.
22. León OS, Martínez G, García I, Bilbao T, Ledesma L. Balance antioxidante/pro-oxidante: salud y enfermedad. La Habana: Eitorial Ciencias Médicas; 2004. p. 2, 61.
23. Fogelman AM, Schechter I, Seager J, Hokom M, Child JS, Edwards PA. Malondialdehyde alteration of low density lipoproteins leads to cholesterol ester accumulation in human monocyte-macrophages. Proc Natl Acad Sci USA. 1980;77:2214-8.
24. Quinn MT, Parthasarathy S, Fong LG, Steinberg D. Oxidatively modified low density lipoproteins: a potential role in recruitment and retention of monocyte/macrophages during atherosclerosis. Proc Natl Acad Sci USA. 1987;84:2995-8.
25. Higashi Y, Sasaki S, Nakagawa K, Kimura M, Noma K, Sasaki S, et al. Excess norepinephrine impairs both endothelium-dependent and _independent vasodilation in patients with pheochromocytoma: a comparison before and after adrenalectomy. Hypertension.2002;39:513-8.
26. Gesquière L, Loreau N, Blache D. Role of the cyclic AMP-dependent pathway in free radical-induced cholesterol accumulation in vascular smooth muscle cells. Free Radic Biol Med 2000;29(2):181-90.
27. Griffiths HR, Aldred S, Dale C, Nakano E, Kitas GD, Grant MG, et al. Homocysteine from endothelial cells promotes LDL nitration and scavenger receptor uptake. Free Radic Biol Med.2006;40:488-500.
28. Larsson H, Böttiger Y, Iuliano L, Diczfaluzy Ulf. In vivo interconversion of 7b
-hydroxycholesterol and 7-ketocholesterol, potential surrogate markers for oxidative stress. Free Radic Biol Med.2007;43:695-701.29. Dzeletovic S, Babiker A, Lund E, Diczfaluzy U. Time course of oxysterol formation during in vitro oxidation of low density lipoprotein. Chem Phys Lipids.1995;78:119-28.
30. Larsson DA, Baird S, Nyhalah JD, Yuan XM, Li W. Oxysterol mixtures, in atheroma-relevant proportions, display synergistic and proapoptotic effects. Free Radic Biol Med.2006;41:902-10.
31. Lizard G. Oxysterol mixtures; a promosing approach to investigate the biological effects of oxysterols: A commentary on "Oxysterol mixtures, in atheroma-relevant proportions, display synergistic and proapoptotic effects. Free Radic Biol Med. 2006;41:872-3.
32. Rosenson RS, Otvos JD, Freedman DS. Relations of lipoprotein subclass levels and low-density lipoprotein size to progression of coronary artery disease in the Pravastatin Limitation of Atherosclerosis in the Coronary Arteries (PLAC-I) trial. Am J Cardiol. 2002;90:89-94.
33. Krauss RM. Heterogeneity of plasma low-density lipoproteins and atherosclerosis risk. Curr Opin Lipidol. 1994;5:339-49.
34. Nichols AV, Krauss RM, Musliner TA. Nondenaturing polyacrylamide gradient gel electrophoresis. Methods Enzymol. 1986;128:417-31.
35. Krauss RM, Burke DJ. Identifi cation of multiple subclases of plasma low density lipoproteins in normal humans. J Lipid Res. 1982;23:97-104.
36. National Cholesterol Education Program (NCEP). Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) fi- nal report. Circulation. 2002;106:3143-421.
37. Austin MA, Rodriguez BL, McKnight B. Low-density lipoprotein particle size, triglycerides, and high density lipoprotein cholesterol as risk factors for coronary heart disease in older Japanese-American men. Am J Cardiol. 2000;86:412-6.
38. Bjornheden T, Babyi A, Bondjers G, Wiklund O. Accumulation of lipoprotein fractions and subfractions in the arterial wall, determined in an in vitro perfusion system. Atherosclerosis 1996; 123:43-56.
39. Weisser B, Locher R, de Graaf J. Low density lipoprotein subfractions increase thromboxane formation in endothelial cells. Biochem Biophys Res Commun 1993; 192:1245-50.
40. Tertov VV, Sobenin IA, Gabbasov ZA. Multipled-modified desialylate low density lipoproteins that cause intracellular lipid accumulation. Isolation, fractionation and characterization. Lab Invest. 1992;67:665-75.
41. Jovinge S, Ares MP, Kallin B, Nilsson J. Human monocytes/macrophages release TNF-alpha in response to Ox-LDL. Arterioscler Thromb Vasc Biol. 1996;16:1573-9.
42. Maziére C, Maziére JC. Activation of transcription factors and gene expression by oxidized low-density lipoprotein. Free Radic Biol Med. 2009;46:127-37.
43. Gu Q, Bowden GT, Normolle D, Sun Y. SAG/ROC2 E3 ligase regulates skin carcinogenesis by stage-dependent targeting of c-Jun/AP1 and IkappaB-alpha/NF-kappaB. J Cell Biol. 2007;1009-23.
44. Rhoades KL, Golub SH, Economou JS. The regulation of of the human of the human tumor necrosis factor alpha promoter region in macrophage, T cell, and B cell lines. J Biol Chem. 1992;267:22102-7.
45. Serkkola E, Hurme M. Synergism between pritein-kinase C and cAMP-dependent pathways in the expression of the interleukin-1 beta gene is mediated via the activator-protein-1 (AP-1) enhancer activity. Eur J Biochem. 1993;213:243-9.
46. Bidder M, Shao JS, Charlton-Kachigian N, Loewy AP, Semenkovich CF, Towler DA. Osteopontin transcription in aortic vascular smooth muscle cells is controlled by glucose-regulated upstream stimulatory factor and activator protein-1 activities. J Biol Chem. 2002;277:44 485-96.
47. Ares MP, Kallin B, Ericksson P, Nilsson J. Oxidized LDL induces transcription factor activator protein-1 but inhibits activation of nuclear factor-kappa B in human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1995;15:1584-90.
48. Maziére C, Djavaheri-Mergny M; Frey-Fresart V, Delattre J, Maziére JC. Copper and cell-oxidized low density lipoprotein induces activator protein-1 in fibroblasts, endothelial and smooth muscle cells. FEBS Lett. 1997;409:351-6.
49. Rivard A, Principe N, Andrés V. Age-dependent increase in c-fos activity and cyclin A expression in vascular smooth muscle cell proliferation and atherosclerosis. Cardiovasc Res. 2000;45:1026-34.
50. Chen Y, Currie RW. Heat shock treatments suppresses angiotensin II-induced SP-1 and AP-1 and stimulate Oct-1 DNA-binding activity in heart. Inflammation Res. 2005;54:338-43.
51. Santamarina-Fojo S, Peterson K, Knapper C, Qiu Y, Freeman L, Cheng JF, et al. Complete genomic sequence of the human ABCA 1 gene: analysis of the human and mouse ATP-binding cassette A promoter. Proc Natl Acad Sci USA. 2000;97:7987-92.
52. Rajavashisth TB, Yamada H, Mishra NK. Transcriptional activation of the macrophage-colony stimulating factor gene by minimally modified LDL: involvement of nuclear factor-kappa B. Arterioscler Thromb Vasc Biol. 1995;15:1591-8.
53. Natarajan R, Reddy MA, Malik KU, Fatima S, Khan BV. Signaling mechanisms of nuclear factor-kappaB-mediated activation of inflammatory genes by 13-hydroperoxy-octadecadienoic acid in cultured vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2001;21:1408-13.
54. De Winther MP, Kanters E, Kraal G, Hofker MH. Nuclear factor kappaB signaling in atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:904-14.
55. Brand K, Page S, Rogler G, Bartsch Brandl R, Knuechel R, Page M, et al. Activated transcription factor nuclear factor-kappaB is present in atherosclerosis lesion. J Clin Invest. 1996;97:1715-22.
56. Ferreira V, van Dijk KW, Groen AK, Vos RM, van der Kaa J, Gijbels MJ, et al. Macrophage-specific inhibition of NF-kappaB activation reduces foam-cell formation. Atherosclerosis. 2007;192:283-90.
57. Chiba T, Kondo Y, Shinozaki S, Kaneko E, Ishigami A, Maruyama M, et al. A selective NF-kappaB inhibitor, DHMEQ, reduced atherosclerosis in ApoE-defficient mice. J Atheroscler Thromb. 2006;13:308-313.
58. Terkeltaub R, Banka CL, Solan J, Santoro D, Brand K, Curtiss LK. Oxidized LDL induces monocytic cell expression of interleukin-8, a chemokine with T-lymphocyte chemotactic activity. Arterioscler Thromb. 1994;14:47-53.
59. Ishii H, Kizaki K, Horie S, Kazama M. Oxidized low density lipoprotein reduces thrombomodulin transcription in cultured human endothelial cells through degradation of the lipoprotein in lysosomes. J Biol Chem. 1996;271:8458-65.
60. White CR, Datta G, Zhang Z, Gupta H, Garber DW, Mishra VK, et al. HDL therapy for cardiovascular diseases: the road to HDL mimetics. Curr Atheroscler Rep. 2008;10:405-12.
61. Barter PJ, Nicholls S, Rye KA. Antiinflammatory properties of HDL. Circ Res 2004; 95: 764-72.
62. Oda MN, Bielicki JK, Ho TT. Paraoxonase 1 overexpression in mice and its effect on high-density lipoproteins. Biochem Biophys Res Commun. 2002;290:921-7.
63. Navab M, Anantharamaiah GM, Reddy ST. Mechanisms of disease: proatherogenic HDL-an evolving field. Nature Clin Pract. 2006;2:504-11.
64. Navab M, Anantharamaiah GM, Fogelman AM. The role of high-density lipoprotein in inflammation. Trends Cardiovasc Med. 2005;15:158-61.
65. Fogelman AM. When good cholesterol goes bad. Nature Med. 2004;10: 902-3.
66. Zhang R, Brennan ML, Fu X. Association between myeloperoxidase levels and risk of coronary artery disease. JAMA. 2001;286:2136-42.
67. Bergt C, Pennathur S, Fu X. The myeloperoxidase product hypochlorous acid oxidizes HDL in the human artery wall and impairs ABCA1-dependent cholesterol transport. Proc Natl Acad Sci USA. 2004;101:13032-7.
68. Ansell BJ, Navab M, Hama S. Inflammatory / antiinflammatory properties of high density lipoprotein distinguish patients from control subjects better than high density lipoprotein cholesterol levels and are favourably affected by simvastatin treatment. Circulation. 2003;108:2751-6.
Recibido: 8 de diciembre de 2009.
Lic. Livan Delgado-Roche. Centro de Estudios para las Investigaciones y Evaluaciones Biológicas, Instituto de Farmacia y Alimentos, Universidad de La Habana. Ave 23 # 21 425 e/ 214 y 222, La Coronela, La Lisa, CP 13 600, La Habana, Cuba. Correo electrónico: livan@cieb.sld.cu

