










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Farmacia
versión On-line ISSN 1561-2988
Rev Cubana Farm v.44 n.4 Ciudad de la Habana oct.-dic. 2010
ARTÍCULOS DE REVISIÓN
Farmacocinética y farmacodinámica, implicación en un uso más racional de los antimicrobianos
Pharmacokinetics and pharmacodynamics: implication in a more rationale use of antimicrobials
Amaury L. Noda AlbeloI; Arturo Vidal TalletII
IEspecialista de I Grado en Inmunología Clínica. Máster en Infectología y Enfermedades Tropicales y en Atención Integral Al niño. Instructor. Hospital Pediátrico Universitario "Eliseo Noel Caamaño". Matanzas, Cuba.
IIEspecialista de II Grado en Pediatría. Máster en Educación Superior y en Enfermedades Infecciosas. Profesor Auxiliar. Hospital Pediátrico Universitario "Eliseo Noel Caamaño". Matanzas, Cuba.
RESUMEN
La farmacodinámica describe la compleja interrelación que se establece entre el perfil farmacocinético del antimicrobiano y la susceptibilidad in vitro de la bacteria. La eficacia clínica y microbiológica del antimicrobiano se puede predecir utilizando 3 parámetros farmacodinámicos: concentración máxima/concentración mínima inhibitoria (Cmax/CMI); área bajo la curva concentración plasmática/tiempo contra CMI (ABC/CMI); duración del intervalo de la dosis que la concentración del antimicrobiano supera la CMI (T> CMI). El tipo particular de antimicrobiano determina qué parámetro es el que mejor predice su eficacia. En la actualidad el uso de los antimicrobianos continúa siendo frustrantemente empírico, sobre todo con respecto al intervalo ínter dosis y el tiempo de duración de la terapia antimicrobiana. Dado que nuevos agentes para patógenos multirresistentes pueden tomar décadas para estar disponibles en el arsenal terapéutico, el conocimiento y aplicación de los principios farmacocinéticos y farmacodinámicos resulta la mejor opción para optimizar la utilización de los viejos y los nuevos antimicrobianos, lo que permite identificar parámetros de exposición a droga íntimamente asociados a la habilidad para destruir microorganismos y suprimir la emergencia de resistencia en subpoblaciones de organismos.
Palabras clave: Farmacocinética, farmacodinámica, antimicrobianos.
ABSTRACT
Pharmadynamics describes the complex interrelation established between the animicrobial pharmacocynetics profile and the in effectiveness of the antimicrobial agent may be predicted using three pharmacodynamics parameters: inhibitory maximal concentration/minimal concentration (Cmax/CMI), area under the curve of plasmatic/time concentration against CMI (ABC/CMI)l lenght of dose interval where concentration pass the CMI (T> CMI). The specific type of the antimicrobial agent determines that the parameter is better po predict its effectiveness. At present time, the use of antimicrobial agent remains being empirically frustrating mainly with respecto to inter-dosis interval and the lenght time of antimicrobial therapy. Given that th new agents for multiresistant pathogens may to go pass decadesto be available in therapeutical arrays, the knowledge and application of pharmacocynetics and pharmacodynamics principles is a better alternative to optimize the use of ancient and new anatimicrobial agents allowint to identify drugs exposition parameters closely related to ability to destroy the microorganisms and to suppress the appearance of resistance in subpopulations of organisms.
Key words: Pharmacocynetics, Pharmacodynamics, antimicrobials.
INTRODUCCIÓN
Los antimicrobianos se encuentran entre las drogas más usadas en la práctica clínica.1 Desafortunadamente en los últimos años la introducción de nuevas drogas antimicrobianas se ha reducido a unos pocos medicamentos, un escenario muy diferente a décadas anteriores, donde la creación o descubrimiento de nuevas sustancias con actividad antimicrobiana era una forma de combatir la resistencia y favorecer el pronóstico de infecciones graves por gérmenes virulentos. Este fenómeno coincide con la explosión en el número de organismos multi-droga-resistentes tanto en la comunidad como en el medio hospitalario.1 En la actualidad el uso de los antimicrobianos continúa siendo frustrantemente empírico, sobre todo con respecto al intervalo ínter dosis y el tiempo de duración de la terapia antimicrobiana. El conocimiento y aplicación de los principios farmacocinéticos y farmacodinámicos es la mejor opción para optimizar la utilización de los viejos y los nuevos antimicrobianos, permitiendo identificar parámetros de exposición a droga íntimamente asociados a la habilidad para destruir microorganismos y suprimir la emergencia de resistencia en sub-poblaciones de organismos.2
FARMACOCINÉTICA
La farmacocinética define la relación que se establece entre el antimicrobiano y el paciente, cómo el organismo manipula la droga, e incluye los procesos de absorción, distribución, unión a proteínas séricas e hísticas, metabolismo y eliminación.3,4 Diferencias en el grado de unión a proteínas séricas pueden originar cambios en la concentración de antibacteriano libre, determinante de la penetración a tejidos y la actividad antibiótica.3
ABSORCIÓN
La absorción de la droga en la circulación sistémica ocurre desde cualquier sitio en el que sea administrada, excepto cuando se administra directamente en un compartimiento fluido fisiológico (fluido cerebrospinal o torrente sanguíneo) donde evidentemente la infusión es directa; esta definición incluye la vía intramuscular, subcutánea, tópica y gastrointestinal después de la administración por vía oral, rectal u otra vía que la lleve al tracto gastrointestinal. La velocidad y el grado de absorción es altamente dependiente de las propiedades físico-químicas de la droga así como del ambiente en el sitio de administración. Propiedades como tamaño molecular, solubilidad, lipofilicidad, estabilidad, influencian la velocidad y extensión de la absorción.5 La cantidad del antimicrobiano que alcanza la circulación sistémica se expresa en porcentaje de la cantidad total que pudo ser adsorbida; este porcentaje se define como biodisponibilidad de la droga.4 Los medicamentos que se adsorben desde el intestino delgado son afectados por el efecto de primer paso por el hígado a través de la circulación portal; drogas administradas por la vía intravenosa, e intramuscular no son afectadas por este fenómeno y tienen mayor biodisponibilidad. La disminución en la perfusión gastrointestinal, subcutánea y muscular que se observa en pacientes sépticos, puede reducir significativamente la absorción de fármacos4,6,7 y por tanto, su biodisponibilidad, generando concentraciones plasmáticas insuficientes de antimicrobianos; en estos pacientes debe priorizarse la administración por vía intravenosa en cuyo caso se alcanza 100 % de absorción.
DISTRIBUCIÓN
La distribución de una droga, es descrita más comúnmente por su volumen de distribución (Vd), el cual no es un volumen real, es un parámetro cinético, permite relacionar la cantidad en el organismo con la concentración en este, es un espacio de dilución.8 Puede definirse como el volumen que debería tener el organismo para que la cantidad presente al equilibrio estuviera a la misma concentración; existen factores que afectan Vd como solubilidad lipídica, coeficiente de partición de la droga entre diferentes tipos de tejidos, flujo sanguíneo en el tejido, pH, y unión a material biológico (ej., proteínas plasmáticas, componentes celulares);4,5 el Vd se puede calcular de la siguiente manera:
![]()
El Vd es variable entre personas, se afecta por factores como, disfunción de órganos excretores u obesidad, y puede tener severas variaciones en un mismo individuo, como consecuencia del aumento de la permeabilidad vascular que acompaña a enfermedades graves, sepsis, quemaduras, cirrosis hepática, insuficiencia cardiaca, renal,4 etc, esto agravado por el aporte masivo de fluidos.5,9,10
Es importante para el clínico el conocimiento del concepto Vd; las drogas con pequeños Vd tienen limitada distribución, mientras que las de grandes Vd se distribuyen extensamente por todo el organismo; de este modo se puede inferir que antimicrobiano, se restringe al espacio intravascular y extravascular, lo cual implica en su distribución el medio intracelular.11,12
El ritmo con que la droga se mueve de la sangre al tejido se describe como aclaramiento distribucional; este describe el volumen de sangre del cual el medicamento es transferido al tejido por unidad de tiempo, el aclaramiento distribuciónal es un proceso bidireccional que refleja el equilibrio en el movimiento de la sangre al tejido y del tejido a la sangre.11
METABOLISMO Y BIOTRANSFORMACIÓN
Los antimicrobianos son metabolizados por reacciones que ocurren en el hígado y en otros órganos. Las reacciones metabólicas son clasificadas como de fase I y fase II. Las reacciones de fase I pueden inactivar, activar o convertir un sustrato activo en otro activo, con mayor, menor o igual actividad; estas reacciones están controladas por el sistema del citocromo P-450; generalmente inactivan al sustrato, lo hacen más polares, lo que facilita su eliminación. Las reacciones de fase II son procesos en los que interviene la conjugación de estos compuestos con grandes moléculas, incluye glucoronidación, sulfatación y acetilación, esto aumenta la polaridad y facilita la excreción.3-5,11
ELIMINACIÓN
La eliminación de sustancias extrañas ocurre por 2 mecanismos fundamentales de excreción, aclaramiento renal (CLr) descrito como el ritmo con que es eliminada una sustancia del organismo a través de los riñones, incluye filtración glomerular, secreción tubular, y difusión pasiva; diferentes antimicrobianos son eliminados por uno o más de estos procesos.4
Aclaramiento no renal, es un término genérico que describe la suma de vías de aclaramiento que no incluyen al riñón; estos mecanismos incluyen vías biliares (ej., ceftriaxone) o intestino (ej., azitromicina); otros mecanismos menos comunes como la eliminación del alcohol a través de la piel y pulmones y la ionización e inactivación de los aminoglucósidos por el esputo y eliminación por la expectoración en fibrosis quística.
El ritmo de aclaramiento renal y no renal se combinan para determinar el ritmo con que es eliminado el antimicrobiano del organismo, esto se conoce como aclaramiento corporal total.11 Del concepto de eliminación deriva el de tiempo de vida media del antimicrobiano (t1/2), la vida media de la droga es el tiempo que se requiere para que la concentración en sangre del compuesto decrezca a la mitad.4 Se considera que la estabilidad en la concentración de una droga se alcanza cuando el paciente la ha estado tomando por un periodo igual a 5 o 7 veces su t1/2 (ej., 5 a 7 días para una droga con una vida media de 24 h); de manera similar se considera que se ha eliminado cuando desde la última dosis ha pasado un tiempo similar a 5 o 7 veces su t1/2; la vida media de una droga varía de paciente a paciente, en ocasiones se reporta como rangos; la unión a proteína y estados de fallos finales de órganos alteran la t1/2 de una droga, Ej. la eliminación de antimicrobianos como beta-lactámicos, vancomicina, aminoglucósidos y quinolonas puede reducirse significativamente en casos de insuficiencia renal, se generan concentraciones plasmáticas más elevadas; en el caso de drogas con margen terapéutico estrecho como aminoglucósidos, puede dar origen a toxicidad, la dosificación de estos fármacos debe ajustarse en forma proporcional a la función renal; los antimicrobianos de eliminación hepática como lincosaminas y antituberculosos deben ajustarse en la disfunción hepática; sin embargo, el aclaramiento de fármacos en casos de insuficiencia hepática es mucho más difícil de estimar. Los antibacterianos que tienen eliminación mixta, como cloxacilina o ceftriaxona, en general no requieren ajuste de dosis frente a la falla de un órgano excretor, por un aumento compensatorio de la depuración por el otro órgano.
Las características de absorción, distribución y eliminación del antimicrobiano determinan la curva concentración/tiempo en plasma, de esta se infiere la concentración que alcanza el fármaco en el tejido infectado, donde, en definitiva se requiere de este en concentraciones adecuadas para el control de la infección. Esta curva define una serie de parámetros farmacocinéticos importantes, concentración máxima (Cmáx), la vida media del antimicrobiano en el plasma (t1/2) y el área bajo la curva (ABC) (fig. 1).
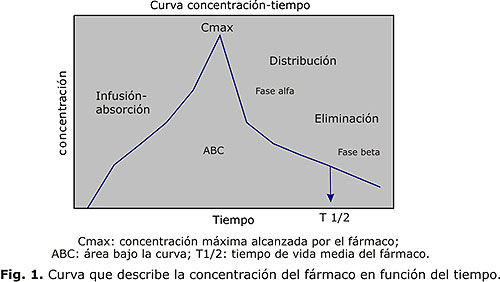
Las concentraciones plasmáticas e hísticas no siempre se correlacionan linealmente, los antibacterianos con menos unión a proteínas como aminoglucósidos y quinolonas, tienen generalmente una correlación plasma/tejidos mayor que los beta-lactámicos que se caracterizan por una elevada unión a proteínas plasmáticas. Ciertas afecciones como meningitis pueden mejorar transitoriamente la penetración de antimicrobianos al sitio infectado, al aumentar la permeabilidad de la barrera hematoencefálica; en general, la penetración a tejidos es relevante en infecciones que afectan órganos con baja penetración de antibacteriano como son el SNC, ojo, huesos, páncreas y pulmón
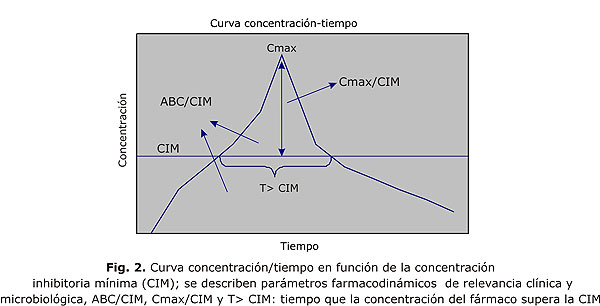
En términos generales, para que un antimicrobiano sea efectivo, debe lograr concentraciones superiores a la concentración inhibitoria mínima del microorganismo (CIM), o sea, para que una bacteria se considere susceptible tiene que tener una CIM alcanzable por el antimicrobiano en su perfil farmacocinético en humanos. La farmacodinámica describe el efecto antimicrobiano del medicamento en el sitio de la infección y también los efectos tóxicos en relación con la concentración de la droga durante la evolución de la terapia.13,14 Los estudios farmacodinámicos se basan en 2 tipos de datos, microbiológicos y farmacocinéticos. La farmacodinámica describe la compleja interrelación que se establece entre el perfil farmacocinético del antimicrobiano y la susceptibilidad in vitro de la bacteria; la curva concentración/tiempo del antimicrobiano se determina en función de CIM (fig. 2). Los parámetros farmacocinéticos son expresados en función de la CIM. El éxito clínico y microbiológico depende de una adecuada interacción farmacodinámica entre el antimicrobiano y la bacteria, lo que permite establecer ciertos objetivos farmacodinámicos en el tratamiento antiinfeccioso, como Cmáx/CIM, ABC/CIM o T> CIM que constituyen demostradamente parámetros predictores de éxito.15
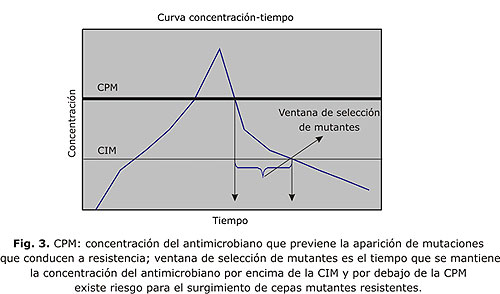
CONCENTRACIÓN DE PREVENCIÓN DE MUTANTES
Concentración de prevención de mutantes (CPM) es un concepto que cada vez es más discutido en la literatura, se refiere a la concentración del antimicrobiano que previene la aparición de mutaciones que conducen a resistencia,16 esta varía para diferentes organismos y para diferentes drogas. De este concepto deriva el de ventana de selección de mutantes, se define como el periodo de exposición del germen al antimicrobiano que se caracteriza por una concentración por debajo de CPM pero por encima de la CIM17-19 (fig. 3); teóricamente en este periodo pueden aparecer gérmenes resistentes al antimicrobiano, obtener niveles superiores a CPM de la droga sería un objetivo durante el tratamiento para evitar la selección de cepas mutantes resistentes.
EFECTO POSANTIBIÓTICO
Durante el testage de antimicrobianos in vitro, se comprobó que al retirar el medicamento existe una demora para que el microorganismo reentre en el periodo logarítmico de crecimiento, este fenómeno se denomina efecto posantibiótico (EPA); la duración de este es dependiente del germen y la droga. Aminoglucosidos, fluoroquinolonas, eritromicina, clindamicina, tetraciclinas y estreptograminias,20 producen in vivo EPA contra organismos gramnegativo, los beta-lactámicos (excepto carbapenémicos) no producen EPA contra organismos gramnegativos;21,22 los beta-lactámicos producen breve EPA contra organismos grampositivos, in vivo fundamentalmente contra estafilococos. De manera general, los antimicrobianos que interfieren con la síntesis proteica o del ADN poseen EPA contra gérmenes gramnegativo y los que interfieren en la síntesis de pared no. Del fenómeno EPA depende el efecto post-antibiótico sub-CIM, se refiere a que el microorganismo en fase de retardo de recrecimiento por el efecto post-antibiótico es más susceptible a reducir su crecimiento a concentraciones sub-CIM y el efecto leucocitario posantibiótico, define que el microorganismo en el estado post-antibiótico de crecimiento es más susceptible a la actividad microbicida leucocitaria. Ambos efectos ocurren in vivo donde la concentración de la droga declina gradualmente en el tiempo. Los mecanismos del EPA son desconocidos, posibles explicaciones incluyen daños no letales inducidos por el medicamento y persistencia del antimicrobiano en el sitio de acción;23 nuevos microorganismos inyectados durante el periodo de EPA, inician un rápido crecimiento sugiriendo que el EPA no es causado por la persistencia de la droga en el tejido. En el orden clínico la presencia o ausencia del EPA se puede usar para variar los esquemas de dosis de los antimicrobianos, un agente con un prolongado EPA se puede usar con menos frecuencia que uno que no posea EPA. Alternativamente, un agente que no posea EPA puede ser más efectivo en infusión continua, de manera que las concentraciones séricas siempre excedan la CIM del germen en cuestión.
El mecanismo de acción de cada familia de antimicrobianos determina una cinética bactericida específica, y la presencia y duración del efecto post-antibiótico. De esta manera se pueden dividir los antimicrobianos en 3 grupos:
• El primer grupo induce muerte concentración dependiente y produce EPA moderado o prolongado.
• En el segundo grupo el patrón de destrucción es dependiente del tiempo de exposición y no produce o tienen un mínimo EPA contra la mayoría de los microorganismos.
• El tercer grupo destruye de manera tiempo dependiente pero difiere del grupo anterior en que poseen EPA moderado o prolongado.
AGENTES QUE DESTRUYEN BACTERIAS DE MANERA CONCENTRACIÓN DEPENDIENTE
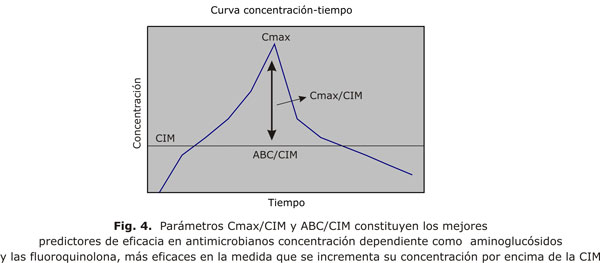
Los antimicrobianos que responden a esta cinética bactericida se caracterizan por ser más eficientes en su actividad a medida que se incrementa su concentración en el sitio de infección, generalmente son óptimos cuando su concentración supera 10 veces la CIM para el germen en cuestión; ejemplos de antimicrobianos con esta dinámica de respuesta son los aminoglucósidos24-26 y las fluoroquinolonas.22,24-27 El parámetro Cmax/CIM (fig. 4), es el mejor predictor de eficacia para este tipo de agente, además in vitro se ha comprobado que es eficaz para predecir el desarrollo de resistencia bacteriana. ABC/CIM también predice eficacia (son co-variables), ABC/CIM (fig. 4), es una medida de la exposición total del germen al antimicrobiano; Cmax/CIM y ABC/CIM son parámetros difíciles de independizar en diseños de ensayos clínicos porque cuando la Cmax/CIM es elevada, usualmente también la ABC/CIM es alta, ambos parámetros son de valor estadístico predictivo e indistinguible desde el punto de vista de cual es de importancia primaria.
AGENTES QUE DESTRUYEN DE MANERA TIEMPO DEPENDIENTE (CONCENTRACIÓN INDEPENDIENTE)
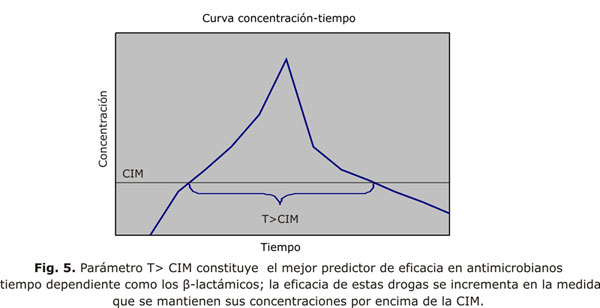
Para estos agentes en general cuando la concentración en el sitio de la infección supera 4 veces la CIM, permiten un nivel de destrucción bacteriana que no se incrementa con concentraciones superiores; el tiempo que se logra concentraciones superiores a la CIM es el que mejor predice la eficacia en esta clase de antimicrobianos; T> CIM, es un parámetro farmacodinámico que indica el periodo de tiempo en que la concentración de un antimicrobiano supera la CIM del germen en cuestión y se da en porcentaje del intervalo de la dosis (fig. 5); existen evidencias que muestran a T> CIM como un predictor farmacodinámico importante en penicilinas, cefalosporinas, carbapenémicos y monobactámicos.24,28-30 La meta en este grupo de medicamento sería prolongar la exposición de la droga. Existe diferencias entre los microorganismos en cuanto al tiempo por encima de la CIM que necesitan para ser adecuadamente eliminados; por ejemplo. penicilinas y cefalosporinas requieren menos tiempo por encima de la CIM para ser eficaces contra estafilococo que contra bacilos gramnegativos y estreptococos. Esto parece deberse a que estafilococo es el único contra el cual estas drogas exhiben "prolongado" EPA. P aeruginosa requiere un tiempo ligeramente mayor por encima de CIM que otros gramnegativo (5 al 10 % del intervalo de la dosis). Para los carbapenémicos existen mínimas diferencias en cuanto a tiempo por encima del CIM requerido para la eficacia contra la mayoría de los bacilos gram-negativos; la mayor diferencia la exhibe el S. pneumoniae con un muy corto tiempo de 10 a un 20 % del intervalo de la dosis para ser eficaz.31-33
El tercer grupo destruye de manera tiempo dependiente pero difiere del grupo anterior en que posee EPA moderado o prolongado; incluye macrólidos, clindamicina, estreptograminas, tetraciclinas, oxazolidinonas, y glicopéptidos.13,22,34,35 La presencia de EPA hace que el tiempo de exposición sea menos importante, la meta serÍa proveer la cantidad adecuada de la droga. El parámetro que mejor predice la eficacia de la droga es ABC/CIM.
IMPORTANCIA CLÍNICA DE ÍNDICES Y PARÁMETROS FARMACODINÁMICOS
Conociendo que la eficacia del antimicrobiano depende del tiempo de exposición, se puede optimizar su eficacia administrándolo de manera que su concentración se mantenga lo más estable posible superando la CIM; se puede esto lograr si se utiliza las siguientes estrategias: 1) administración en infusión continua; 2) intervalos ínter dosis más cortos, aunque con concentraciones más bajas; o 3) sustituyéndolo por un antimicrobiano de mayor T1/2. El objetivo final es lograr porcentajes elevados de T> CIM.
En antimicrobianos como los aminoglucósidos en los que la eficacia depende de la concentración, independientemente del tiempo que se supere la CIM, será necesario administrar dosis más elevadas, aunque en intervalos ínter dosis más largos; de este modo se optimiza su utilización y el objetivo es lograr altos Cmáx/CIM y ABC/CIM, parámetros predictores de éxito en este grupo de medicamentos.
Por otro lado, los antimicrobianos con un EPA prolongado podrían ser administrados en intervalos aun más largos sin temor a perder eficacia, ya que los microorganismos no recrecerían durante un lapso de tiempo aunque los niveles fueran muy bajos.
Todos estos valores de predicción de actividad y parámetros asociados, como el EPA, CPM tiene una aplicación clínica muy bien definida: el diseño y optimización de las pautas de dosificación de los antimicrobianos y a largo plazo prevenir la aparición de resistencia.
ASPECTOS QUE SE DEBEN CONSIDERAR EN LA VALORACIÓN DE PARÁMETROS FARMACOCINÉTICOS/FARMACODINÁMICOS
Actualmente los parámetros farmacocinéticos/farmacodinámicos para agentes antimicrobianos se sustentan en la concentración plasmática y la CIM in vitro; el uso de la concentración total plasmática y los valores in vitro de CIM no es lo ideal. El tejido no es un compartimiento homogéneo, la distribución de la droga en plasma y tejido depende de sus características físico-químicas, el tiempo de equilibrio tiene un rango desde minutos hasta días. Implica el área de superficie capilar, el volumen de fluido en el compartimiento místico y la afectación de la distribución por las barreras anatómicas barrera hemato/encefálica (BHE), ojos, próstata, hueso; algunos antimicrobianos se unen de manera importante a proteínas hísticas o celulares. Es común creer que la concentración del antibiótico libre en equilibrio de concentración en plasma y en fluido hístico es igual, basados en el precepto de que la fuerza de distribución es la difusión pasiva; de este modo se trata de predecir la concentración hística infiriéndola de la plasmática, pero esto no siempre es correcto; la diferencia entre la concentración total del plasma y la concentración libre en tejido puede ser significativa en innumerables situaciones, como en drogas de alta unión a proteínas, y la penetración en los tejidos de difícil acceso. Todo esto hace que la concentración plasmática total no sea un valor de salida ideal farmacocinético para definir dosis de medicamento. La penetración a tejidos depende de variables como difusión, transporte activo, liposolubilidad, y unión a proteínas; por ejemplo, en infecciones del sistema nervioso central, los antimicrobianos lipofílicos no ionizados como rifampicina y metronidazol penetran ampliamente, mientras que la mayoría de los beta-lactámicos, quinolonas y glicopéptidos tiene una penetración limitada y requieren ser administrados en dosis máximas que se puede ver favorecida por el aumento de permeabilidad que acompaña a la infección, los aminoglucósidos y las cefalosporinas de 1ª y 2ª generación tienen mínima penetración. En osteomielitis la penetración del antimicrobiano también es clave para el éxito de la terapia, las lincosaminas tienen alta penetración; vancomicina y quinolonas logran concentraciones superiores a la CIM de los principales patógenos. Solo la concentración de la droga libre en el sitio diana es responsable del efecto terapéutico, por lo que el parámetro farmacocinético ideal para el análisis farmacocinético/farmacodinamico sería la concentración de la droga libre en el fluido intersticial.
LA APLICACIÓN DE LOS PRINCIPIOS FARMACODINÁMICOS AL PACIENTE CRÍTICO
La aplicación de los principios farmacodinámicos en el paciente grave es complicado por los cambios farmacocinéticos que se operan en estos pacientes, el incremento en el Vd, decrecimiento en las concentraciones de proteínas séricas, decrecimiento en el metabolismo y aclaramiento de la droga (disfunción de órgano o hipo perfusión), incremento del metabolismo y aclaramiento por el estado hipermetabólico del paciente crítico.36 A pesar de los cambios inherentes al paciente crítico, la optimización de las dosis basadas en la caracterización farmacocinética de este y la apropiada aplicación de los principios farmacodinámicos ofrecen un potencial indiscutible para mejorar la sobrevida del paciente y para prevenir la emergencia de resistencia. Por la severidad de la infección en el paciente crítico y las variabilidades farmacocinéticas, así como las relacionadas con la penetración en tejido, la recomendación general para la dosis de antimicrobianos son estrategias de dosis agresivas; bajas dosis de antimicrobianos pueden fallar para erradicar el patógeno y predisponen al desarrollo de resistencia. El uso de altas dosis de medicamento potencialmente compensan las alteraciones farmacocinéticas presentes, lo que incrementa la posibilidad de alcanzar las metas farmacodinámicas adecuadas que se correlacionan con éxito terapéutico, pero no se debe perder la perspectiva del incremento en las reacciones
adversas.
La farmacodinamia permite al clínico seleccionar la droga más potente y ofrece una guía para definir la dosis e intervalos de dosis más inocuos y eficaces para un patógeno particular en un sitio específico de infección. Para la industria farmacéutica la aplicación de estos principios ayuda a predecir las probabilidades de éxito del compuesto en desarrollo y guía a la hora de definir regímenes de dosis en el diseño de ensayos clínicos. Dado que la introducción de nuevos agentes antimicrobianos para patógenos multi-droga-resistentes puede tomar décadas para estar disponibles en el arsenal terapéutico, la opción más inmediata es optimizar el uso de los que se tienen a nuestra disposición.
REFERENCIAS BIBLIOGRÁFICAS
1. Drusano GL. Pharmacokinetics and pharmacodynamics of antimicrobials. Clin Infect Dis. 2007;45(Suppl 1):89-95.
2. Rybak MJ. Pharmacodynamics: relation to antimicrobial resistance. Am J Med. 2006; 6(Suppl 1):37-44.
3. Bergman SJ. Pharmacokinetic and pharmacodynamic aspects of antibiotic use in high-risk populations. Infect Dis Clin North Am. 2007;21(3):821-46.
4. Levison ME, Levison JH. Pharmacokinetics and Pharmacodynamics of Antibacterial Agents. Infect Dis Clin North Am. 2009;23(4);791-815.
5. Boucher BA. Pharmacokinetic changes in critical illness. Crit Care Clin. 2006;22(2):255-71.
6. Singh G, Chaudry KI, Chudler LC. Sepsis produces early depression of gut absorptive capacity: restoration with diltiazem treatment. Am J Physiol. 1992;263:19-23.
7. Johnston JD, Harvey CJ, Menzies IS. Gastrointestinal permeability and absorptive capacity in sepsis. Crit Care Med. 1996;24:1144-9.
8. Benet LZ, Kroetz DL, Sheiner LB. Pharmacokinetics: The dynamics of drug absorption, distribution, and elimination In: Hardman JG, Limbird LE, ed. Goodman and Gilman's the Pharmacological Basis of Therapeutic. 9th ed. New York: McGraw-Hill; 1996. p. 3-28.
9. Ronchera-Oms CL, Tormo C, Ordovas JP. Expanded gentamicin volume of distribution in critically ill adult patients receiving total parenteral nutrition. J Clin Pharm Ther. 1995;20:253-8.
10. Etzel JV, Nafziger AN, Bertino JS. Variation in the pharmacokinetics of gentamicin and tobramycin in patients with pleural effusions and hypoalbuminemia. Antimicrob Agents Chemother. 1992;(36):679-81.
11. Mandell GL, Douglas RG, Bennett JE. Mandell, Douglas, and Bennett's. Principles and practice of infectious diseases. [cover art] 7th:2 v. (xxxviii, 3661, cxxx p.). Available from: http://www.mdconsult.com/book/player /book.do?method=display&type=aboutPage&decorator=header&eid=4-u1.0-B978-0-443-06839-3..X0001-X—TOP&isbn=978-0-443-06839-3&uniq=216473411#lpState=open&lpTab=contentsTab&content=4-u1.0-B978-0-443-06839-3..00020-5%3Bfrom%3Dtoc%3Btype%3DbookPage%3Bisbn%3D978-0-443-06839-3
12. Quintiliani R. Pharmacokinetics/Pharmacodynamics for critical care clinicians. Crit Care Clin. 2008;24(2):335-48.
13. Craig WA. Basic pharmacodynamics of antibacterials with clinical applications to the use of Beta-lactams, glycopeptides, and linezolid. Infect Dis Clin North Am. 2003;(17):479-501.
14. Roberts JA, Lipman J. Antibacterial dosing in intensive care: Pharmacokinetics, degree of disease and pharmacodynamics of sepsis. Clin Pharmacokinet. 2006; (45):755-73.
15. Roberts JA, Kruger P, Paterson DL. Antibiotic resistance-What's dosing got to do with it? Crit Care Med. 2008;(36):2433-40.
16. Zhao X, Drlica K. Restricting the selection of antibiotic-resistant mutants: A general strategy derived from fluoroquinolone studies. Clin Infect Dis. 2001;33 (Suppl 3):147-56.
17. Olofsson SK, Marcusson LL. Selection of ciprofloxacin resistance in Escherichia coli in an in vitro kinetic model: Relation between drug exposure and mutant prevention concentration. J Antimicrob Chemother. 2006;(57):1116-21.
18. Knudsen JD, Odenholt I, Erlendsdottir H. Selection of resistant Streptococcus pneumoniae during penicillin treatment in vitro and in three animal models. Antimicrob Agents Chemother. 2003;(47):2499-506
19. Firsov AA, Smirnova MV, Lubenko IY. Testing the mutant selection window hypothesis with Staphylococcus aureus exposed to daptomycin and vancomycin in an in vitro dynamic model. J Antimicrob Chemother. 2006;(58):1185-92.
20. Ling TK. In vitro activity and post-antibiotic effect of quinupristin/dalfopristin (Synercid). Chemotherapy. 2001;47(4):243-9.
21. Lortholary O, Tod M, Cohen Y, Petitjean O. Aminoglycosides. Med Clin North Am. 1995;(79):761-87.
22. Roberts JA, Kruger P, Paterson DL. Antibiotic resistance-What's dosing got to do with it? Crit Care Med. 2008;(36):2433-40.
23. Stubbings W. Mechanisms of the post-antibiotic effects induced by rifampicin and gentamicin in Escherichia coli. J Antimicrob Chemother. 2006;58(2):444-8.
24. Lortholary O, Lefort A, Tod M. Pharmacodynamics and pharmacokinetics of antibacterial drugs in the management of febrile neutropenia. Lancet Infect Dis. 2008;( 8):612-20.
25. Tam VH. Pharmacodynamic modeling of aminoglycosides against Pseudomonas aeruginosa and Acinetobacter baumannii: identifying dosing regimens to suppress resistance development. Antimicrob Agents Chemother. 2008;52(11):3987-93.
26. Mouton JW, Vinks AA. Pharmacokinetic/pharmacodynamic modelling of antibacterials in vitro and in vivo using bacterial growth and kill kinetics: The minimum inhibitory concentration versus stationary concentration. Clin Pharmacokinet. 2005;(44):201-10.
27. Tam VH, Louie A, Deziel MR, et al. Bacterialpopulation responses to drug-selective pressure: Examination of garenoxacin's effect on Pseudomonas aeruginosa. J Infect Dis. 2005;(192):420-8.
28. Angus BJ, Smith MD, Suputtamongkol Y. Pharmacokinetic-pharmacodynamic evaluation of ceftazidime continuous infusion vs intermittent bolus injection in septicaemic melioidosis. Br J Clin Pharmacol. 2000;(50):184-91.
29. Ikawa K. Pharmacodynamic evaluation of biapenem in peritoneal fluid using population pharmacokinetic modelling and Monte Carlo simulation. Int J Antimicrob Agents. 2008;32(4):339-43.
30. Drusano GL. Antimicrobial pharmacodynamics. Critical interactions of bug and drug. Nat Rev Microbiol. 2004;(2):289-300.
31. Roberts JA, Paratz J, Paratz E. Continuous infusion of beta-lactam antibiotics in severe infections: A review of its role. Int J Antimicrob Agents. 2007;(30):11-8.
32. Jaruratanasirikul S, Sriwiriyajan S, Punyo J. Comparison of the pharmacodynamics of meropenem in patients with ventilatorassociated pneumonia following administration by 3-hour infusion or bolus injection. Antimicrob Agents Chemother. 2005;(49):1337-9.
33. Drusano GL, Sorgel F, Quinn J. Impact of pharmacodynamic dosing of meropenem on emergence of resistance during treatment of ventilator-associated pneumonia: A prospective clinical trial. In: Interscience Conference of Antimicrobial Agents and Chemotherapy: 16-19/12/2005. Washington: American Society for Microbiology; 2005.
34. Moise-Broder PA, Forrest A, Birmingham M. Pharmacodynamics of vancomycin and other antimicrobials in patients with Staphylococcus aureus lower respiratory tract infections. Clin Pharmacokinet. 2004;(43):925-42.
35. Bowker KE. pharmacodynamics of minocycline against Staphylococcus aureus in an in vitro pharmacokinetic model. Antimicrob Agents Chemother. 2008;52(12):4370-3.
36. Scaglione F. Pharmacokinetics/pharmacodynamics of antibacterials in the Intensive Care Unit: setting appropriate dosing regimens. Int J Antimicrob Agents. 2008;32(4):294-301.
Recibido: 8 de junio de 2010.
Aprobado: 17 de julio de 2010.
Dr. Amaury L. Noda Albelo. Hospital Pediátrico Universitario "Eliseo Noel Caamaño".Matanzas, Cuba. Correo electrónico: amauryn.mtz@infomed.sld.cu


{kind=link}
{kind=link}
{kind=link}