










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Farmacia
versión On-line ISSN 1561-2988
Rev Cubana Farm v.44 n.4 Ciudad de la Habana oct.-dic. 2010
ARTÍCULOS DE REVISIÓN
Las Buenas Prácticas en la Producción de Biológicos y los Sistemas de Gestión de la Calidad
The Good Practices in the Production of biological agents and the Quality Mangement Systems
Marisel Guadalupe Quintana EsquivelI; Isabel Apezteguía RodríguezII
IDoctora en Ciencias Biológicas. Centro de Ingeniería Genética y Biotecnología, La Habana, Cuba.
IIMáster en Tecnología y Control de Medicamentos. Investigadora Agregada. Centro de Ingeniería Genética y Biotecnología, La Habana, Cuba.
RESUMEN
En este trabajo se presenta un análisis de los aspectos reguladores relacionados con las Buenas Prácticas de Fabricación, en particular en la producción de biológicos y biotecnológicos, y se especifican los requerimientos complementarios que se deben cumplir en estos procesos. Se describen los rasgos distintivos de la industria biotecnológica, destacándose su variabilidad intrínseca debido a su origen, tipo de proceso y el uso de ensayos biológicos. Se introducen los nuevos paradigmas de la industria farmacéutica en el siglo XXI, incorporándose el uso de las técnicas modernas de gestión tipo ISO 9000 y su armonización con las Buenas Prácticas de Fabricación. Se describen las herramientas actuales para llevar a cabo este objetivo. Se plantea que implementar un sistema de calidad farmacéutico según la International Conference of Harmonization (ICH) Q10 basado en estos criterios, permite seguir disminuyendo los riesgos asociados en la fabricación de medicamentos, aumentar la eficiencia y lograr mayores éxitos dentro y fuera de la organización.
Palabras clave: Buenas Prácticas de Fabricación, sistema de gestión de la calidad, sistema de calidad farmacéutico.
ABSTRACT
In present paper an analysis of regulators related to the Good Practices of manufacture, particularly in the biological and biotechnological methods specifying the complementary requierements fulfilled in these processes. Distinctive characteristic of biotehnological industry enphasizing its intrinsic variability due to its origin, the type of process and the use of biological trials. At XXI new paradigms of pharmaceutical industry are introduced adding the use of current management techniques type ISO 9000 and its harmonization with the Good Practices of manufacture. The curent tools are described to achieve this objetive. Authors propose to apply a system of pharmaceutical qulity according to the International Conference of Harmonization (ICH) Q10 on the base of these criteria allows to decrease the risks associated with drugs manufacture, to increase the efficiency and to achieve more success inside and ouside of the organization.
Key words: Good Practices of Manufacture, quality management system, pharmaceutical cuality system.
INTRODUCCIÓN
Los productos biológicos, incluyendo los biotecnológicos, son sustancias que se pueden extraer de fuentes naturales como tejidos o fluidos, órganos de humanos, animales y plantas, del crecimiento de agentes microbianos y virus, incluyendo los obtenidos por la tecnología del ADN recombinante y los anticuerpos monoclonales.
Gracias al desarrollo de la genética y la biología molecular es posible introducir genes de una especie en otra; las células microbianas y de organismos superiores modificados genéticamente son capaces de expresar en fermentadores de gran volumen, los productos de interés para el hombre en las cantidades y condiciones de pureza y seguridad necesarias. Entre las diferencias de los productos biotecnológicos modernos y los productos biológicos convencionales están el alto porcentaje de pureza de su ingrediente farmacéutico activo (IFA), así como la caracterización físico-química y biológica.1 Esta tecnología ha permitido la producción de moléculas terapéuticas y profilácticas, que en algunos casos han sustituido otras que anteriormente eran obtenidas a partir de tejidos humanos o animales, por ejemplo la hormona del crecimiento o la insulina.2
A diferencia de los medicamentos farmacéuticos tradicionales, que normalmente se fabrican y controlan usando técnicas físico-químicas reproducibles, los productos biológicos se obtienen con métodos que involucran procesos y materiales biológicos, los cuales tienen una variabilidad intrínseca, es decir el rango y naturaleza de los subproductos son variables. Por esta razón, en la obtención de biológicos es necesario apegarse cabalmente a las Buenas Prácticas de Fabricación (BPF) en todos los pasos del proceso, incluyendo la fabricación del IFA.3-5
En la actualidad la industria farmacéutica y biotecnológica necesita incorporar los principios de los sistemas de calidad modernos, armonizados con las BPF, con el objetivo de minimizar los riesgos asociados a la fabricación de medicamentos e incrementar la incorporación de avances tecnológicos y aumentar su eficiencia.
En este trabajo se presentan los aspectos reguladores asociados a las BPF en la producción de biológicos/biotecnológicos, las características de esta industria y las tendencias hacia un sistema de gestión de calidad moderno, el cual llevará a tener una mayor seguridad y eficiencia en nuestras producciones.
ASPECTOS REGULADORES EN LA FABRICACIÓN DE PRODUCTOS BIOLÓGICOS
Las BPF se definen como: "conjunto de lineamientos y actividades relacionadas entre sí, destinadas a garantizar que los productos farmacéuticos elaborados, tengan y mantengan la identidad, pureza, concentración, potencia e inocuidad requerida para su uso".6 Entre los principios básicos de las BPF se puede citar: la organización y el personal, las instalaciones y el equipamiento, los insumos y su control, la producción y el control de proceso, el aseguramiento de la calidad en la producción y en los laboratorios analíticos, la contratación de servicios productivos y de ensayo, así como cumplir con la calificación y validación en equipos, procesos y sistemas.
Las BPF es la norma básica que plantea los aspectos esenciales que deben cumplirse en las plantas de medicamentos para la obtención de productos seguros y eficaces. A partir de esta, se han derivado regulaciones relacionadas con procesos particulares como son las Buenas Prácticas (BP) en la producción de biológicos, complementarias a las BPF farmacéuticas y donde se plantean algunos requerimientos específicos.3-5,7 Recientemente la norma Europea: "The rules governing medicinal products in the European Union" , Volume 4 "Good Manufacturing Practice: medicinal products for human and veterinay use" en su Draft Annex 2 "Manufacture of biological medicinal products for human use" 2008, se amplían los principios de estas BP y se presentan los nuevos productos biológicos como: los derivados de la terapia génica, la terapia celular somática y productos de plantas y animales transgénicos.5
Teniendo en cuenta lo planteado en estas regulaciones (cuadro), se muestran los aspectos básicos en la aplicación de las BPF en biológicos, los cuales están relacionados con las características de estos procesos y productos, donde la experiencia y capacitación del personal, la importancia del control del proceso, las características del control de calidad y la disminución de los contaminantes indeseables, constituyen elementos esenciales para el logro de estas producciones.
Cuadro. Aspectos básicos en la aplicación de las Buenas Prácticas
| | Cumplimiento de las Buenas Prácticas en Biológicos |
| Persona | - Tener experiencia en la fabricación de biológicos y capacitación en higiene, microbiología, virología, inmunología, veterinaria. |
| Instalaciones y Equipamiento | - Contar con un programa de monitoreo ambiental acorde con el producto y a la etapa del proceso, incluyendo los métodos de detección de microorganismos específicos. |
| Documentación | - Para materiales biológicos se requiere información sobre su fuente, origen, método de fabricación y ensayos aplicados, particularmente los controles microbiológicos. |
| Materiales* | - Procedentes de proveedores evaluados y liberados por control de calidad. |
| Producción y su control | - Control de los bancos celulares y de su estabilidad, procedimientos aprobados. - Determinar la carga microbiana durante las etapas de fermentación y purificación, necesario para prevenir o minimizar contaminantes indeseables, asociados a enzimas bacterianas, endotoxinas, etc. - Establecer los límites de aceptación en dependencia de los requerimientos del producto y la capacidad del proceso para remover o inactivar los contaminantes. |
| Control de Calidad | - Llevar a cabo controles esenciales para la calidad de estos productos, como la remoción del virus, contenido del ADN residual, contenido de proteínas contaminante del hospedero, los cuales no pueden efectuarse en el producto terminado y deben chequearse en una etapa apropiada del proceso. |
Otra normativa de gran aplicación en la industria farmacéutica y biotecnológica es la referida a las BPF para la producción de productos estériles, incorporando conceptos de clasificación de áreas limpias, así como límites para el monitoreo ambiental.9-11 También se han emitido otras guías/normativas importantes para los fabricantes de biofarmacéuticos, como son las BP en la producción del IFA, la cual ha sido publicada por la ICH: del inglés "International Conference Harmonization" (Conferencia Internacional de Armonización) y adoptada por las agencias reguladoras, FDA: del inglés "Food and Drug Administration" (Administración de Drogas y Alimentos) y EMEA: del inglés "The European Agency for the Evaluation of Medicinal Products" (Agencia Europea de Medicamentos).12-14
Rasgos distintivos en la fabricación de biológicos/biotecnológicos
•Es una industria altamente regulada, basada en el cumplimiento de guías y normativas de diferentes organismos como son: Centro para el Control Estatal de la Calidad de los Medicamentos (CECMED), Organización Mundial de la Salud (OMS), ICH, FDA, EMEA. Estas reglamentaciones están relacionadas con los aspectos que se deben garantizar en la industria para cumplir con las BPF, buenas prácticas en los laboratorios de control y en las buenas prácticas clínicas, entre otros.
•Requiere que el personal tenga un alto nivel profesional y técnico, el cual se enfrenta a tecnologías novedosas basadas en aspectos científico-técnicos que involucra diferentes áreas del conocimiento: Genética, Microbiología; Bioquímica, Química, Ingeniería, Ciencias Farmacéuticas, Automatización, entre otros.
•Existe un alto nivel de inversiones. Para poder llevar a cabo la fabricación de estos productos se debe contar con instalaciones clasificadas, con sistemas de aire, agua y vapor, así como el equipamiento adecuado que cumplan las etapas de calificación y validación. Además, deben cumplir con las mejoras tecnológicas que le impone el estado del arte en esta industria.
•Variabilidad intrínseca de los procesos. Estos productos se obtienen a partir de materiales biológicos, lo cual introduce al proceso y al control una mayor variabilidad. De ahí la necesidad de disponer de un control estadístico y el cumplimiento de los puntos críticos del proceso, además de mantener el estado validado de estos. Es necesario un conocimiento profundo de la sustancia activa y su fórmula farmacéutica, y que se cumpla con los atributos de calidad del producto.
•Requiere de un gran número de animales. En el caso de las pruebas de experimentación y control de calidad que están relacionadas con los ensayos biológicos (potencia y seguridad) se necesitan animales, por ejemplo, curieles, ratones, conejos, que deben mantenerse bajo seguimiento de salud y en instalaciones adecuadas para este trabajo. Desde hace algunos años se trabaja en la sustitución y la correlación de las técnicas biológicas por otros métodos analíticos, con vistas a disminuir el número de animales a utilizar en el control de calidad de estas producciones.15
•Requiere de la liberación lote a lote para la comercialización. El establecimiento por parte de las agencias reguladoras de efectuar a liberación lote a lote, obligatoria para las vacunas y los productos derivados de la sangre y eventualmente para otras formas farmacéuticas, es propia de la producción de biológicos. Esta forma de trabajo ha constituido una vía de disminuir los riesgos propios de la producción de estos medicamentos.16
SISTEMAS DE CALIDAD. LA FAMILIA ISO 9000:2000
Los expertos en este tema, expusieron sus criterios acerca del concepto de calidad: "adecuación al uso", según Joseph M. Juran; "conformidad con los requisitos establecidos; ausencia de defectos; imperfecciones o contaminación; satisfacción del cliente", según Philip B. Crosby, referido por Padilla.17 Otras definiciones fueron adoptadas por regiones y países: "la totalidad de las características de una entidad que influyen en su capacidad para satisfacer necesidades establecidas e implícitas".18 "Capacidad de un conjunto de características inherentes de un producto, sistema o proceso para satisfacer los requisitos de los clientes y otras partes interesadas".19 Todas las definiciones son válidas, desde el concepto de calidad relacionado con el producto, sus características y el cliente hasta la definida en la ISO 9000:2000 que tiene en cuenta no solo el producto y el cliente sino también toda la organización, describiéndose el sistema de gestión de la calidad .
Para conocer los principios de los sistemas de gestión, fue necesario contar con un organismo encargado de la armonización de normas técnicas en diferentes campos. Su nombre, en inglés es: International Organization for Standardization, sus fundadores eligieron la sigla ISO para darse a conocer, por ser corta y universal. Su función principal es buscar la estandarización de normas de productos y seguridad para las empresas u organizaciones a escala internacional. Está constituido por los miembros de órganos nacionales de normalización de más de 160 países, sistema voluntario, no gubernamental con sede en Suiza. En Cuba este órgano lo representa la Oficina Nacional de Normalización.
La familia ISO 9000:2000 es un conjunto de guías internacionales genéricas que establecen sistemas de gestión de la calidad aplicados a organizaciones de cualquier tipo o tamaño. La norma ISO 9001:2000,20 específica los requisitos para un sistema de gestión de la calidad, cuando la organización necesita demostrar su capacidad para proporcionar, de forma coherente productos que satisfagan los requisitos acorde con su uso y reglamentarios aplicables, y aspira a aumentar la satisfacción del cliente a través de la aplicación eficaz de este sistema. Recientemente ha sido aprobada la ISO 9001:2008.21 que profundiza algunos puntos relacionados con los procesos de contratación a externos, las características del representante de la dirección para el sistema de gestión, así como una nueva guía sobre los diferentes métodos de medición y seguimiento en la satisfacción del cliente. Además, aumenta la compatibilidad con la norma ISO 14001:200422 relacionada con el medio ambiente.
ARMONIZACIÓN ENTRE LOS SISTEMAS DE GESTIÓN DE LA CALIDAD Y LAS BUENAS PRÁCTICAS DE FABRICACIÓN VIGENTES EN LA INDUSTRIA FARMACÉUTICA
Con la emisión por parte de la FDA del documento "Pharmaceutical GMP's for the 21st Century. A risk based approach" en el año 2004,23 se dan a conocer las dificultades presentes en esta industria, basado entre otras, en su poca eficiencia y falta de incorporación de nuevas tecnologías, planteándose la necesidad de incorporar técnicas modernas de gestión con vistas a ganar en eficiencia y seguridad. A partir de esta situación la mayoría de las autoridades reguladoras, bajo las normas de las buenas prácticas de fabricación vigentes (BPFv), recomiendan a los fabricantes de medicamentos diseñar un sistema de aseguramiento de la calidad o de gestión de la calidad, indicando que nos encontramos en un tránsito hacia la armonización entre los sistemas de gestión de la calidad tipo ISO 9001:2000 y las BPF características de esta industria.
Para una mejor comprensión de este tema, se analizará lo que aparece en diferentes regulaciones sobre este aspecto:
-La regulación No. 16-2006 del CECMED plantea: "El fabricante establecerá y mantendrá un sistema de gestión de la calidad basado en los requisitos generales de la ISO 9001:2000, donde se incluirán los requisitos específicos de las BPF de productos farmacéuticos vigentes, el que será plenamente documentado y su eficacia monitoreada".6
-La serie de informes técnicos No. 908, 2003 de la OMS plantea: "Para alcanzar el objetivo de calidad en la fabricación de medicamentos, es decir productos seguros y eficaces, hay que diseñar e implementar un sistema de aseguramiento de la calidad (SAC) que incorpore las BPF y el control de calidad, así como otras acciones asociadas. Este sistema debe contar con personal, instalaciones, equipos, procesos y materiales adecuados. La alta dirección de la organización tiene la responsabilidad por el desarrollo e implantación de este sistema. Sus elementos deben ser documentados y debe verificarse sistemáticamente su efectividad".24
-La Agencia Española de Medicamentos y Productos Sanitarios, en la Guía de normas de correcta fabricación de medicamentos de uso humano y veterinario. Parte 1, capitulo1: Gestión de calidad del 2008 plantea "Los medicamentos producidos deben cumplir con el registro sanitario autorizado para la comercialización. El cumplimiento de los objetivos de calidad es responsabilidad de la alta directiva y requieren de la participación y el compromiso del personal a todos los niveles de la compañía, sus proveedores y distribuidores. Se debe diseñar un SAC.25
Como se aprecia en las recomendaciones dadas por las agencias reguladoras, los productores de medicamentos deben diseñar e implantar un sistema de calidad, ya sea de gestión o de aseguramiento, donde se incorporen los aspectos de las normas tipo ISO y el cumplimiento de las regulaciones de las BPF.
Estos sistemas, como muestra la figura, ya sea de aseguramiento o de gestión, deben cumplir con los requisitos de las BPF y el control de la calidad, y adicionar como herramientas de dirección: la revisión anual de producto y el análisis de riesgo.25 Además, incorporar nuevos conceptos de los sistemas de gestión como la responsabilidad de la alta gerencia en los objetivos de calidad, la eficiencia y eficacia de los procesos y sistemas.
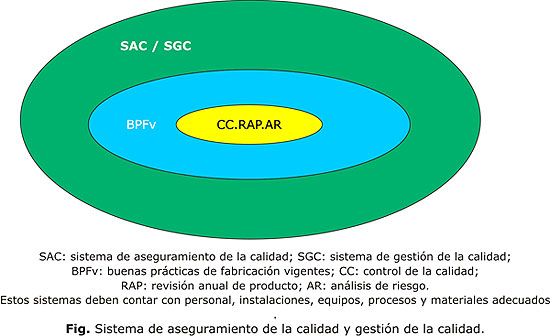
SISTEMA DE CALIDAD FARMACÉUTICO
La industria farmacéutica y biotecnológica comienza a incorporar la filosofía de los sistemas de calidad modernos armonizados con los principios de las BPFv. Desde finales del siglo pasado, la FDA ha planteado la necesidad de cambiar paradigmas, entre los que se encuentran, incorporar el análisis de riesgo y los modelos de gestión de calidad modernos, aspectos donde esta industria ha llegado tarde en relación con otras.23 Para cumplir con estos objetivos la comunidad productiva y reguladora se ha propuesto desarrollar herramientas que ayuden a la interpretación y el cumplimiento de los nuevos desafíos, basándose en el empleo de documentos aprobados y adoptados recientemente como los citados a continuación:
- ICH Q8 "Pharmaceutical Development", noviembre 2005.26
- ICH Q8 (R1) "Pharmaceutical Development", noviembre 2008.27
- ICH Q9 "Risk Management ", noviembre 2005.28
- ICH Q10 "Pharmaceutical Quality System", junio 2008.29
Otra norma de la FDA "Guidance for Industry Quality Systems Approach to Pharmaceutical Current Good Manufacturing Practice Regulations", September 2006,30 describe un modelo de gestión comprensible, bajo los requerimientos de las BPFv, para la producción de medicamentos de uso humano y veterinario, así como los biológicos. Está relacionada con la aplicación de los sistemas de calidad en la industria farmacéutica y ha servido de ayuda a los organismos reguladores y fabricantes para la comprensión y aprobación de la ICH Q10, en junio del 2008.
La implementación de los principios que aparecen en esta trilogía (Q8, Q9 y Q10), supone una revolución en el modo de trabajo de las empresas farmacéuticas que opten por esta forma de organización. Es sin duda un reto, que llevará a otro modo de funcionamiento y logrará productos más seguros y de organizaciones más eficaces con claras ventajas competitivas.31,32
La ICH Q10, es un modelo para lograr una adecuada administración del sistema de calidad farmacéutico (SCF) durante el ciclo de vida del producto (desarrollo farmacéutico, transferencia de tecnología, fabricación y discontinuidad del producto). Se aplica a las sustancias activas y productos farmacéuticos, incluyendo los biológicos y biotecnológicos, sin pretender crear ninguna expectativa adicional más allá de los requerimientos regulatorios actuales.29
Los sistemas de calidad incorporan nuevas formas de trabajo orientadas a la satisfacción del cliente, ampliando este concepto hacia otros ámbitos, administrar con una visión preventiva, fomentar el desarrollo del personal, liderar la organización con un enfoque a costos, disminuyendo las perdidas por no calidad e implementar la mejora continua. Esta según la ISO 9000:2000 constituye "la acción recurrente que aumenta la capacidad para cumplir los requisitos". Su implementación en el SCF permitirá aumentar la calidad en el producto, reducir la variabilidad del proceso, introducir las innovaciones y lograr una mayor rentabilidad.
En estos sistemas, cada empresa debe establecer los indicadores que permitan evaluar la calidad y eficiencia de sus actividades y procesos. Son conocidos como KPI'S: del inglés "Key Performance Indicador" (indicadores clave del desempeño). Estos servirán de retroalimentación que permitan valorar los avances y retrocesos, comunicar el estado de situación y evolución a todos los niveles de la organización, tanto interno como externo. Es recomendable que la organización establezca indicadores al menos de sus procesos estratégicos y clave.
¿QUÉ SE GANA CON LA IMPLEMENTACIÓN DE LOS SISTEMAS DE CALIDAD FARMACÉUTICO?
La armonización de los sistemas de calidad, modelo ISO 9000 y las regulaciones de las BPFv con la implantación del SCF, según la ICH Q10 presenta las siguientes ventajas:
- Incremento de la función de la alta gerencia ante los objetivos de la calidad.
- Incorporación de los indicadores del desempeño como medida de la eficiencia del sistema.
- Logro de la satisfacción del cliente (interno y externo).
- Obtención de un mayor beneficio económico y reconocimiento internacional para la empresa.
La implementación de un sistema de gestión de la calidad en la industria farmacéutica y biotecnológica, manteniendo los niveles de calidad de los medicamentos aumentará la seguridad y la eficiencia en nuestras producciones, y propiciará un ambiente de éxito en toda la organización de la empresa.
REFERENCIAS BIBLIOGRÁFICAS
1. United States Pharmacopoeial Convention. The National Formulary (NF 25). USP XXXI: United States Pharmacopoeia. 31 ed. Rockville: Mack Printing; 2008. p. 409-19.
2. Timón M, Ruiz S. Bases regulatorias de los productos de origen biotecnológico. Economía de la Salud. 2007;6(6):346-51.
3. WHO. Good manufacturing practices for biological products. In: WHO Expert Committee on Specifications for pharmaceutical preparations. Thirty_third Report. Geneva: Word Health Organization; 1993. p. 20-36. (WHO Technical Report Series, No. 834).
4. Annex 2. Manufacture of biological medicinal products for use human. In: The rules governing medicinal products in the European Union. Volume 4. EU Guideline to Good manufacturing practices: medicinal products for human and veterinary use. Brussels: European Commission; 2003. p. 77-82.
5. Draft Annex 2. Manufacture of biological medicinal products for human use. In: The rules governing medicinal products in the European Union. Volume 4. EU Guideline to GMP: medicinal products for human and veterinary use. Brussels: European Commission; 2008. p. 1-25.
6. Regulación No. 16-2006 Directrices sobre las Buenas Prácticas de Fabricación de productos farmacéuticos. La Habana: CECMED. 2006 .p. 1-56.
7. Tabera J. Novedades en las normas de correcta fabricación: Fabricación de medicamentos biológicos para uso humano. FarmEspaña Industrial. 2008:mayo-junio;58-61.
8. WHO. Guidelines on Transmissible Spongiform Encephalopathies in relation to Biological and Pharmaceutical Products. Geneva: Word Health Organization; 2003. p.1-25.
9. Annex 1. Manufacture of sterile medicinal products. In: The rules governing medicinal products in the European Union. Volume 4. EU Guideline to GMP: medicinal products for human and veterinary use. Brussels: European Commission; 2009. p. 1-16.
10. Anexo 4. Producción de productos estériles. En: Regulación 16-2000. Directrices sobre Buenas Prácticas de Fabricación de Productos Farmacéuticos. La Habana: CECMED; 2000. p. 1-24.
11. WHO. Good Manufacturing Practices for sterile pharmaceutical products. In: WHO Expert Committee on Specifications for pharmaceutical preparations. Thirty_sixth Report. Geneva: Word Health Organization; 2002. p. 76-93. (Technical Report Series, No. 902).
12. International Conference of Harmonization (ICH) Q7. Good manufacturing practice. Guide for active pharmaceutical ingredient. Step 4, november, 2000. p. 1-43.
13. Guidance for Industry. Q7A GMP. Guidance for active pharmaceutical ingredient. International Conference of Harmonization (ICH), FDA, august, 2001. p. 1-51.
14. Part II. Basic requirements for active substances used as starting materials. In: The rules governing medicinal products in the European Union. Volume 4. EU Guidelines to GMP: medicinal products for human and veterinary use. Brussels: European Commission; 2005. p. 1-56.
15. Martínez Y, Rodríguez O, Vázquez M, García G, Izquierdo M, Costa L, et al. Correlation of in vivo-in vitro potency assays for the Cuban Hepatitis B Vaccine. Biotecnología Aplicada. 2005;22(1):34-6.
16. Regulación 19-2006. Requisitos y procedimientos para la liberación de lotes de productos biológicos. La Habana: CECMED; 2006. p. 1-10.
17. Padilla G. Autores de la Gestión de la Calidad. 2002. Disponible en: http://www.monografias.com/trabajos10/gesca/gesca.shtml?monosearch
18. COPANT-ISO 8402:1995 Gestión de la Calidad y Aseguramiento de la Calidad. Fundamentos y vocabulario. ISO, Ginebra, 1995 p. 1-60.
19. ISO 9000:2000 Sistema de gestión de la calidad. Fundamentos y vocabulario. ISO, Ginebra, 2000. p. 1-32.
20. ISO 9001:2000 Sistemas de gestión de la calidad. Requisitos. ISO, Ginebra, 2000. p.1-24.
21. ISO 9001:2008 Sistema de gestión de la calidad. Requisitos. (ISO, Ginebra, 2008) p. 1-31.
22. ISO 14001:2004 Sistemas de gestión ambiental. Requisitos con orientación para su uso. (ISO, Ginebra, 2004. p. 1-26.
23. Pharmaceutical cGMPs for the 21st Century-A Risk-Based Approach. FDA, september 2004. p. 1-26.
24. WHO. Good manufacturing Practice for Pharmaceutical: main principles. In: WHO Expert Committee on Specifications for pharmaceutical preparations. Thirty-seventh Report. Geneva: Word Health Organization; 2003. p. 36-89. (Technical Report Series, No. 908).
25. Guía de normas de correcta fabricación de medicamentos de uso humano y veterinario. Parte 1. Capitulo 1. Gestión de Calidad. En: Agencia Española de Medicamentos y Productos sanitarios. 2008. p. 1-5. Disponible en: http://ec.europa.eu/atoz_en.htm
26. International Conference of Harmonization (ICH) Q8 Pharmaceutical Development. Step 5, november 2005:1-7. Available from: http://www.pmda.go.jp/ich/q/q8_06_9_1e.pdf
27. International Conference of Harmonization (ICH) Q8 (R1) Pharmaceutical Development. Step 4, november 2008:1-24. Disponible en: http://www.ich.org/LOB/media/MEDIA4986.pdf
28. International Conference of Harmonization (ICH) Q9 Risk Management. Step 4, november 2005: 1-19. Available from: http://www.ema.europa.eu/Inspections/docs /ICHQ9Step4QRM.pdf
29. International Conference of Harmonization (ICH) Q10 Pharmaceutical Quality System. Step 4, June 2008:1-17. Available from: http://www.ich.org/LOB/media/MEDIA3917.pdf
30. Guidance for Industry. Quality Systems Approach to Pharmaceutical Current Good Manufacturing Practice Regulations. FDA, september, 2006. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation /Guidances/UCM070337.pdf
31. Geijo F, Ruiz A, Tébar A. Desarrollo y registro farmacéutico después de ICH Q8, Q9, Q10: el horizonte del sector. FarmEspaña Industrial. 2006: marzo/abril;2-6.
32. Tazón F. La Consultoría 2.0 en la Nueva Industria Farmacéutica. FarmEspaña Industrial. 2007:julio/agosto;44-50.
Recibido: 8 de junio de 2010.
Aprobado: 17 de julio de 2010.
Dra. Marisel Guadalupe Quintana Esquivel. Centro de Ingeniería Genética y Biotecnología. Ave.31 e/ 158 y 190, Cubanacán, Playa, PO Box 6162, La Habana 10 600, Cuba. Correo electrónico: marisel.quintana@cigb.edu.cu

