










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Farmacia
versión impresa ISSN 0034-7515
Rev Cubana Farm vol.45 no.3 Ciudad de la Habana jul.-set. 2011
ARTÍCULO ORIGINAL
Validación de métodos analíticos para el control de calidad de naproxeno supositorios
Validation of analytical methods for the quality control of Naproxen suppositories
Yaslenis Rodríguez HernándezI, Yania Suárez PérezII, Oscar García PulpeiroIII, Orestes Yuniel Hernández ContrerasIV
I Licenciada Ciencias Farmacéuticas. Máster en Ciencias, Tecnología y Control de Medicamentos. Profesor Instructor. Instituto de Farmacia y Alimentos. Universidad de la Habana. La Habana, Cuba.
II Doctor en Ciencias Farmacéuticas. Licenciada en Ciencias Farmacéuticas. Profesor Titular. Instituto de Farmacia y Alimentos. Universidad de la Habana. La Habana, Cuba.
III Licenciado en Ciencias Farmacéuticas. Máster en Ciencias, Tecnología y Control de Medicamentos. Empresa Laboratorio "Roberto Escudero Díaz". La Habana, Cuba.
IV Licenciado en Ciencias Farmacéuticas. Laboratorios Medsol. La Habana, Cuba.
RESUMEN
En el presente trabajo se desarrollaron por primera vez, los métodos de análisis que serán utilizados para el control de calidad de las futuras formulaciones de supositorios de naproxeno, para uso infantil y adulto de producción nacional. Se propuso un método por espectrofotometría ultravioleta directa, el cual resultó específico, lineal, exacto y preciso para su aplicación en el control de calidad del naproxeno en supositorios, teniendo en cuenta la presencia de grupos cromóforos en su estructura. Se modificó el método por volumetría ácido-base semiacuosa directa reportado para control de calidad de la materia prima de naproxeno y se adaptó al control de calidad en los supositorios. A partir del proceso de validación realizado, se demostró la adecuada especificidad frente a los componentes de la formulación, así como su linealidad, exactitud y precisión en el rango de 1 a 3 mg/mL. Se compararon los resultados obtenidos por ambos métodos sin detectar diferencias estadísticamente significativas entre las réplicas analizadas por cada dosis, por lo que cualquiera de ellos pueden aplicarse al control de calidad de los supositorios.
Palabras clave: naproxeno, volumetría, espectrofotometría ultravioleta, control de calidad, validación, supositorios.
ABSTRACT
The analysis methods that will be used for the quality control of the future Cuban-made Naproxen suppositories for adults and children were developed for the first time in this paper. One method based on direct ultraviolet spectrophotometry was put forward, which proved to be specific, linear, accurate and precise for the quality control of Naproxen suppositories, taking into account the presence of chromophore groups in their structure. Likewise, the direct semi-aqueous acid-base volumetry method aimed at the quality control of the Naproxen raw material was changed and adapted to the quality control of suppositories. On the basis of the validation process, there was demonstrated the adequate specificity of this method with respect to the formulation components, as well as its linearity, accuracy and precision in 1-3 mg/ml range. The final results were compared and no significant statistical differences among the replicas per each dose were found in both methods; therefore, both may be used in the quality control of Naproxen suppositories.
Key words: Naproxen, volumetry, ultraviolet spectrophotometry, quality control, validation, suppositories.
INTRODUCCIÓN
El naproxeno constituyeun poderoso agente antiinflamatorio, utilizado también como analgésico y antipirético, por lo que resulta de gran uso en la terapéutica. Es un derivado del ácido propiónico, compuesto que surge ante la aparición de reacciones adversas del ácido acetil salicílico como irritante de la mucosa gástrica.1,2
El naproxeno se ha empleado con éxito en diferentes formas farmacéuticas. En el mundo se comercializa en forma de gel, crema, suspensión oral, inyectable, sobres, cápsulas, comprimidos y supositorios.3-8 En Cuba, solo se produce naproxeno en forma de tabletas.9
El desarrollo de naproxeno en supositorios para uso infantil y adulto, constituye una línea de investigación priorizada de la Empresa "Roberto Escudero Díaz", único laboratorio productor en Cuba de esta forma farmacéutica. Los supositorios son medicamentos de elección en determinados grupos poblacionales, atendiendo a los efectos indeseables de la administración por la vía oral, de los analgésicos antiinflamatorios no esteroidales (AINEs). Actualmente solo están registrados en Cuba los supositorios de piroxicam, fármaco que ejerce similar efecto que el naproxeno, pero provoca gran variedad de reacciones adversas, por lo que se ha limitado su uso.
Como parte del conjunto de investigaciones que se requieren para el registro de un nuevo producto, se encuentran los estudios analíticos y de validación. Por esta razón el objetivo de este trabajo fue establecer los métodos de análisis que serán utilizados para el control de calidad de las futuras formulaciones de naproxeno para uso infantil y adulto.
MÉTODOS
Desarrollo de métodos para control de calidad de naproxeno en supositorios
Espectrofotometría UV directa
Se desarrolló un método por espectrofotometría UV mediante un procedimiento directo. En la etapa de desarrollo del método se empleó placebo cargado.
Determinación de la longitud de onda de máxima absorción del naproxeno en solución
En la etapa de desarrollo del método se determinó la longitud de onda (l) de máxima absorción del naproxeno materia prima en solución. Se preparó una solución de concentración 0,02 mg/mL, empleando una mezcla cloroformo: etanol (50:50) como disolvente, a la cual se le determinó el espectro UV en el rango de 200 a 400 nm.
Procesamiento de la muestra para el análisis
Teniendo en cuenta que el método se diseñó para el control de calidad del naproxeno en supositorios, fue necesario proponer una metodología analítica que permitiera obtener una solución del principio activo de concentración adecuada a partir de la forma terminada:
A continuación se describe el método propuesto:
1. Pesar individualmente en balanza analítica 10 supositorios.
2. Calcular el peso promedio.
3. Fundir los supositorios hasta obtener una masa homogénea.
4. Agitar homogenizando hasta enfriar la masa.
5. Pesar una masa equivalente al contenido de naproxeno que se desea analizar (100 %), según composición de la formulación.
6. Trasvasar cuantitativamente la base previamente pesada a un volumétrico de 100 mL con el analito.
7. Añadir una mezcla de cloroformo: etanol (50:50).
8. Agitar hasta disolver y enrasar.
9. Tomar una alícuota de 4 mL.
10. Trasvasar a un volumétrico de 50 mL.
11. Completar a volumen con la mezcla de cloroformo: etanol (50:50).
Volumetría ácido-base semiacuosa directa
Se adaptó la volumetría ácido-base semiacuosa directa propuesta en la Farmacopea Británica (BP) del 2009 para control de calidad de naproxeno materia prima al control de calidad de naproxeno en supositorios. Se realizaron algunos cambios en el método y en el tratamiento de la muestra. En la etapa de desarrollo del método se empleó placebo cargado.
Descripción del procedimiento
Teniendo en cuenta que el método se destina a la cuantificación de naproxeno en una matriz diferente, se diseñó un procedimiento sencillo previo a la cuantificación, el cual se describe a continuación:
1. Pesar una cantidad de naproxeno equivalente a 200 mg, y asumir este valor como el 100 % y la cantidad de grasa dura correspondiente.
2. Añadir 75 mL de etanol y 25 mL de H2O destilada.
3. Agitar vigorosamente y tapar el frasco Erlenmeyer.
4. Calentar en baño de María con agitación constante a una temperatura de 50 ± 5 °C hasta la completa fusión de la grasa dura.
5. Agitar vigorosamente y dejar enfriar hasta que la grasa dura comience a solidificar nuevamente.
6. Añadir 3 gotas de fenoftaleína y valorar con NaOH 0,1 mol/L.
Validación de los métodos para control de calidad
Tanto el método espectrofotométrico como el volumétrico, fueron validados según los parámetros exigidos para la categoría I5 que incluye las técnicas destinadas a cuantificar principios activos en las formas terminadas. Los parámetros analizados fueron:
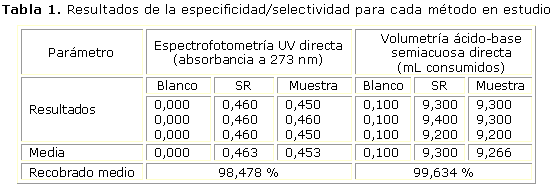
Especificidad/selectividadSe evaluó por triplicado placebos del producto, solución de referencia (SR) y blanco. Se procesaron por triplicado placebos del producto mediante cada método propuesto y se compararon los resultados obtenidos con los resultantes del análisis de placebos cargados con naproxeno en cantidades equivalentes al 100 %.
- Espectrofotometría UV directa: la respuesta obtenida fue la absorbancia a 273 nm y el espectro de absorción UV de 200 a 400 nm.
- Volumetría ácido-base semiacuosa directa: la respuesta obtenida fue mL de valorante consumidos.
Se determinó la posible interferencia de los componentes de la matriz a través de la comparación entre la respuesta obtenida por triplicado para el placebo, el blanco y la SR de naproxeno a la concentración equivalente al 100 %. Se calculó la respuesta media para cada matriz y el recobrado medio total.Criterio de aceptación: no deben aparecer señales analíticas para el placebo en el rango de interés analítico para el naproxeno.
Linealidad del métodoLa linealidad del método, tanto en el método espectrofotométrico como en el volumétrico se realizó analizando 5 concentraciones de naproxeno SR por triplicado, en un rango de 50 a 150 % de la cantidad teórica declarada como 100 %. Se prepararon placebos de cada producto, los cuales se cargaron con las alícuotas correspondientes de SR.
En ambos casos se construyó una curva de calibración de respuesta analítica (Y) vs. concentración teórica expresada en % (X). Los resultados se procesaron estadísticamente mediante el programa STATGRAPHICS versión 5.1 (opción regresión lineal múltiple) y se determinó: r (coeficiente de correlación lineal), r2 (coeficiente de determinación), a (intercepto) y b (pendiente), para el 99 % de confianza. Además se calcularon los factores de respuesta (¦), el valor medio y el CV.
Criterios de aceptación:
- Ecuación de la recta: y= bx + a
- r³ 0,99
- r2³ 0,98
- Prueba de proporcionalidad del método analítico o hipótesis nula de la ordenada en el origen a= 0.
- Se empleó la prueba estadística de la t de Student para n-2 grados de libertad, siendo n el número total de valores donde: texp < ttab.
- Prueba de la hipótesis nula de la pendiente: b= 0. Se determinó a partir de una prueba ANOVA de la regresión, o mejor, la probabilidad asociada al valor de la pendiente, es decir, si la p<< 0,05, el valor de "b" difiere significativamente de cero.
- El coeficiente de variación de los factores de respuesta (CVf) debe ser menor que 5 %.
ExactitudSe construyeron las curvas de recuperación de % recuperado (Y) vs. % teórico (X) de los puntos equivalentes al 50, 100 y 150 % analizando por triplicado placebos cargados por cada método. Los resultados fueron procesados estadísticamente de igual forma que la curva de calibración de la linealidad del método. Además se calculó el % de recobro (R), el recobrado medio (
) y del coeficiente de variación total (CV).
Criterios de aceptación:
CV£ 3,0 %
Además, se realizó la prueba G de Cochran, para determinar si el factor concentración tiene alguna influencia en los resultados. El valor de Gexp se comparó con el de Gtab (µ= 0,05; k= 3; n= 3).
Donde:
k: grupos experimentales.
n: determinaciones por grupo.
Si Gexp< Gtab las varianzas de las 3 concentraciones son equivalentes, o lo que es igual, el factor concentración no influye en la variabilidad de los resultados.
Por último se aplicó la prueba de la t de Student para demostrar que no existen diferencias significativas entre el valor medio de recobro obtenido y el 100 %.
PrecisiónRepetibilidad
Para la repetibilidad se evaluaron por triplicado placebos cargados con la concentración equivalente al 100 % y un valor bajo y otro alto comprendido dentro del rango de la linealidad del método. Se calculó el CV en estos 3 niveles de concentración y se comparó con el criterio establecido. Las determinaciones las realizó el mismo analista con las mismas condiciones de trabajo.
Criterio de aceptación: CV£ 3,0 %.
Precisión intermediaParticiparon 2 analistas, en 2 días diferentes, en el mismo laboratorio. Se analizaron por triplicado en cada caso muestras equivalentes al 100 % del ingrediente farmacéutico activo (IFA). Se calculó el CV total.
Criterio de aceptación: CV< 30 %.
Adicionalmente se realizaron otras pruebas para evaluar la influencia del analista y el día en los resultados analíticos, estas fueron:
- Prueba F de Snedecor: se utilizó para determinar si existían diferencias significativas entre los resultados de los analistas que emplearon el mismo método y entre los días en que se realizaron los análisis. Se calculó el valor de Fexp y se comparó con Ftab para a= 0,05, F1= n-1 grados de libertad del numerador y F2= n-1 grados de libertad del denominador. Si Fexp< Ftab no existe diferencia significativa entre la precisión alcanzada por los analistas, en 2 días de trabajo.
- Prueba de la t de Student: se utilizó para comprobar si los valores obtenidos entre los analistas, que emplearon igual método y los 2 días en que realizaron los análisis eran homogéneos, para el nivel de significación µ= 0,05 y los grados de libertad seleccionados ¦= (n1+n2)-2.
El valor de texp se comparó con el valor de la ttab para a= 0,05 y ¦= n1 + n2 - 2.
Rango: se estableció el intervalo en que se cumplieron satisfactoriamente los criterios de linealidad, exactitud y precisión.
Comparación de los resultados obtenidos por ambos métodos
Una vez validados ambos métodos, se aplicaron por triplicado al control de calidad de supositorios de naproxeno 50 y 500 mg de dosis, correspondientes al uso infantil y adulto respectivamente. Los resultados se compararon estadísticamente aplicando análisis de varianza. Se utilizó el programa STATGRAPHICS versión 5.1 Plus.
RESULTADOS
Espectrofotometría UV directa
La concentración prefijada en el procedimiento propuesto como 100 % fue de 0,02 mg/mL.
El espectro UV del naproxeno, SR, en cloroformo: etanol (50:50), se muestra en la figura 1 (A). Se obtuvieron 4 máximos de absorción al observar el comportamiento en cloroformo:etanol (50:50). Se seleccionó para la cuantificación la lmáxima= 27 nm.
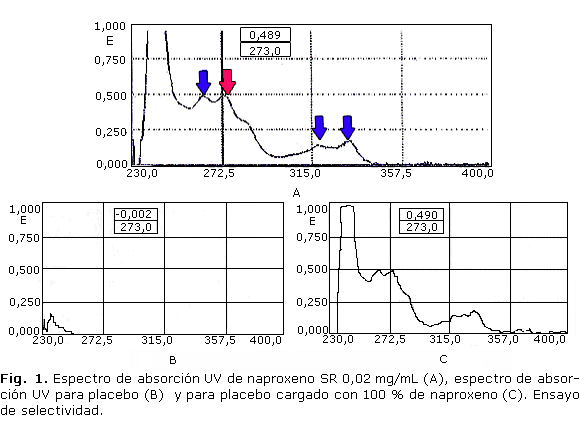
Una vez seleccionada la l de máxima absorción con la concentración de fármaco cuya absorbancia fue adecuada (concentración de 0,02 mg/mL con valores de absorbancia alrededor de 0,489), se procedió a establecer una metodología analítica muy sencilla que permitiera obtener una solución transparente a partir de la forma terminada.
Volumetría ácido-base semiacuosa directa
La concentración establecida como 100 % se correspondió con la reportada en la BP 2009 para la materia prima. Los disolventes se mantuvieron en la misma proporción, sustituyendo el metanol por etanol.
Resultados de la validación de los métodos para control de calidad
El primer parámetro analizado durante la etapa de validación fue la especificidad. En la figura 1 (B) se muestra el espectro UV del placebo tratado por el método propuesto. Como se observa, el placebo no absorbió a 273 nm, valor seleccionado como la l de máxima absorción para la cuantificación del naproxeno, tal como se mostró en el espectro UV de la SR (Fig. 1) (A), el cual coincide perfectamente con la respuesta del placebo cargado con 100 % de analito (Fig. 1) (C).
Se obtuvieron los mismos resultados para blancos y placebos en cada método (tabla 1), lo cual significó que no existieron interferencias en la respuesta analítica debido a la presencia de la base que se empleó en la formulación de supositorios. Por esta razón, no fue necesario aplicar el análisis de varianza.
Estos resultados demuestran la selectividad del método frente a los restantes componentes de la formulación.
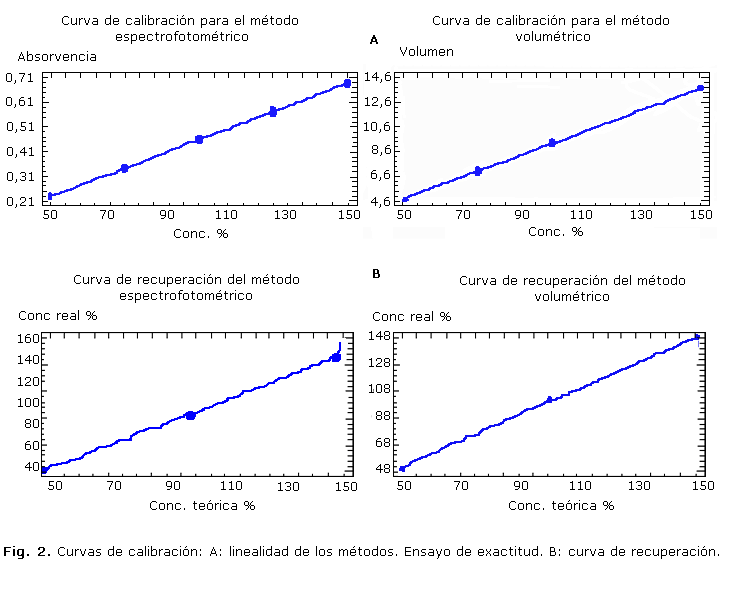
Los resultados de la linealidad para ambos métodos, se muestran en la tabla 2. La linealidad en ambos métodos fue satisfactoria según el cumplimiento de los parámetros estadísticos establecidos.
Al realizar el procesamiento estadístico de los resultados de la exactitud, se comprobó el cumplimiento de todos los criterios de aceptación exigidos, tal como aparece en la tabla 2. La curva de recuperación correspondiente se presenta en la figura 2 (B). Ambos métodos fueron exactos y no se afectaron por errores sistemáticos de forma significativa.
Para la repetibilidad, en ambos métodos, se estimó el CV para 3 niveles de concentración: bajo, medio y alto. Se reportaron C.V bajos, inferiores al 1,5 % establecido como límite (tabla 2).
La precisión intermedia también fue evaluada y dio resultados satisfactorios. Se obtuvieron en ambos casos CV acorde con el criterio de aceptación (CV£ 3,0 %). Se cumplieron satisfactoriamente las restantes pruebas aplicadas, los errores aleatorios no repercutieron en los métodos desarrollados.
El conjunto de resultados obtenidos permitió establecer como rangos para cada método los siguientes:
- Método espectrofotométrico: 0,01-0,03 mg/mL.
- Método volumétrico: 1,00-3,00 mg/mL.
Comparación de los resultados obtenidos por ambos métodos
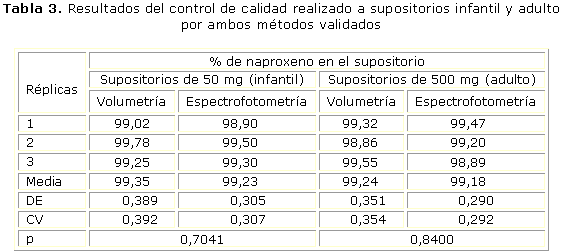
En la tabla 3 se resumen los resultados obtenidos para supositorios de uso infantil y adulto una vez aplicados los métodos validados para control de calidad. Además se refleja el valor de p obtenido al aplicar análisis de varianza a los resultados obtenidos con ambos métodos para supositorios de igual dosis.
DISCUSIÓN
Espectrofotometría UV directa
Se desarrolló un método espectrofotométrico directo para el control de calidad del naproxeno en supositorios, ya que se cuenta con el equipamiento necesario y además es un método ventajoso por su rapidez y sencillez; se propuso un procedimiento analítico desarrollado en el laboratorio para el control de calidad del naproxeno en supositorios. Su aplicación se fundamenta por la presencia de grupos cromóforos en la estructura de este compuesto.
Se decidió aplicar un procedimiento directo, debido a que la composición de la formulación solo incluye IFA y la base del supositorio, la cual no debe afectar los resultados analíticos (no presenta grupos cromóforos), este procedimiento resultó el más sencillo.
Se tomó como referencia la concentración utilizada en la BP 2009 para la identificación por UV del naproxeno en supositorios y suspensión oral. La concentración prefijada en el procedimiento propuesto como 100 % fue de 0,02 mg/mL en correspondencia con la adecuada proporción IFA-base en las futuras formulaciones de naproxeno supositorios.
Para el tratamiento de la muestra previo al análisis se consideraron las características de la matriz en estudio, por lo que se propuso el procedimiento habitual correspondiente a los supositorios tipo suspensión. La agitación de la masa fue el paso crítico para evitar la posible deshomogenización del analito durante el enfriamiento, por su insolubilidad en el Rosupol.
En las pruebas de identificación por UV se utilizó como disolvente el metanol. Sin embargo, se propuso la mezcla cloroformo:etanol (50:50), considerando la necesidad de incluir un disolvente adecuado para la base (cloroformo), mientras que el IFA resultó igualmente soluble en etanol y metanol. La selección de etanol se justifica teniendo en cuenta su menor costo y mayor disponibilidad. Se comprobó experimentalmente que tanto las cantidades utilizadas de naproxeno como de base correspondiente al 100% fueron totalmente solubles en 100 mL de esta mezcla.
Teniendo en cuenta lo reportado en la bibliografía para este IFA, existen 4 posibles máximos de absorción en metanol: 262, 271, 316 y 331 nm respectivamente. Al observar el comportamiento en cloroformo: etanol (50:50), se obtuvo un desplazamiento batocrómico. Esto se debe al efecto del disolvente que está relacionado con el grado de interacción soluto-disolvente, el cual será mayor a mayor polaridad del disolvente. La solvatación altera los niveles electrónicos del cromóforo por la presencia de electrones no pareados.
La l máxima seleccionada fue 273 nm, ya que en la esta se alcanzaron valores adecuados de absorbancia (absorbancia= 0,49) próximos al ideal para la cuantificación: 0,5. Además se encuentra más alejada de la zona de absorción de la mayoría de los disolventes orgánicos.
Volumetría ácido-base semiacuosa directa
La volumetría ácido-base semiacuosa directa es el método reportado en la BP del 2009 para la cuantificación de naproxeno materia prima. Este método se puede utilizar por la presencia en la estructura del grupo carboxilo libre que le confiere características de ácido fuerte al compuesto. El método se nombró «semiacuoso» debido a que fue preciso disolver el analito en una disolución etanol: agua (75:25), ya que el naproxeno es insoluble en agua. Por ser un método sencillo, rápido y fácilmente aplicable en cualquier laboratorio de análisis, se propone su aplicación al control de calidad del naproxeno supositorios. Para ello fue preciso establecer un tratamiento previo teniendo en cuenta las características de los excipientes de la forma terminada.
La concentración establecida como 100 % se correspondió con la reportada en la BP 2009 para la materia prima. Los disolventes se mantuvieron en la misma proporción, sustituyendo el metanol por etanol, se tuvieron en cuenta las mismas razones expuestas con anterioridad en el método espectrofotométrico.
Como parte de los nuevos pasos relacionados con las características de la matriz, se sugirió el calentamiento como coadyuvante a la etapa de disolución, con el objetivo de facilitar la liberación del IFA desde la base del supositorio. Como el naproxeno es insoluble en la base, el enfriamiento requirió de agitación hasta solidificación, para garantizar que el analito se encontrara libre en el medio.
Validación de los métodos para control de calidad
Para la espectrofotometría UV directa se utilizó el término selectividad, pues como se mostró en el espectro UV del placebo (Fig. 1) (B), sí existió absorción a las l más bajas por ser una zona que no provocó interferencia al método.
Para la volumetría ácido-base semiacuosa directa resultómás apropiado el término "especificidad", ya que la respuesta analítica solo se presentó ante un grupo analíticamente activo: el carboxilo libre, presente únicamente en el analito.
El procesamiento estadístico de los datos de la linealidad para ambos métodos (tabla 2), dio resultados satisfactorios, y se demostró el cumplimiento de todos los criterios de aceptación establecidos.
Las ecuaciones de regresión en los 2 casos, presentaron elevados coeficientes de correlación lineal y de determinación. Estos resultados avalan la proporcionalidad existente entre la respuesta analítica y la concentración del analito en el rango analizado, para ambos métodos.
Los coeficientes de recobrado medio quedaron dentro del límite permitido (97-103 %) y los CV totales para cada caso, fueron inferiores al 3,0 % establecido. Adicionalmente se evaluó la influencia de la concentración de analito en la varianza (S) de los resultados a través de la prueba de G de Cochran (tabla 2). Como la Gexp< Gtab las varianzas de los 3 niveles de concentración evaluados por ambos métodos, fueron equivalentes. Es decir, no influyó el factor con centración en la exactitud de ambos métodos. Por su parte, la prueba de la t de Student corroboró la exactitud, ya que no existieron diferencias estadísticamente significativas entre el recobrado medio y el 100 %.
En el estudio de la precisión, el análisis se complementó con las pruebas de F de Snedecor y t de Student. En ambos análisis la Fexp< Ftab en los 2 métodos estudiados, por lo que no existieron diferencias significativas entre las precisiones de los analistas, independientemente del día en que se efectuó el ensayo. Los valores de las texp resultaron menores que las ttab en cada caso, para cada uno de los métodos en estudio, por lo que no hubo diferencias significativas entre los valores medios obtenidos por los analistas. El conjunto de estos resultados permitió asegurar que estos fueron homogéneos y ratificaron la precisión de los métodos en estudio.
En los intervalos utilizados se garantizó adecuada exactitud y precisión, pues los puntos extremos (50 y 150 %) fueron los seleccionados como niveles bajo y alto para las determinaciones realizadas. Teniendo en cuenta la mayor sensibilidad del método espectrofotométrico, se logró un rango que incluyó concentraciones menores con respecto al del método volumétrico, el cual estuvo limitado por la sensibilidad de la balanza utilizada.
El cumplimiento de las exigencias establecidas para la validación de técnicas analíticas, garantizó que los métodos desarrollados, fueran suficientemente específicos, lineales, precisos y exactos para la cuantificación del naproxeno en supositorios, por lo que son ambos métodos válidos para realizar el control de rutina de este nuevo medicamento, en el rango estudiado en cada caso.
Los resultados de la comparación de ambos métodos por triplicado a supositorios de igual dosis, permitió afirmar que no existen diferencias estadísticamente significativas. El análisis de varianza de los datos se aplicó entre grupos (métodos) y dentro de cada grupo (réplicas para supositorios de igual dosis analizados con el mismo método). Para supositorios de 50 mg de dosis para uso infantil se obtuvo un valor de p para la prueba F de 0,7041, el cual es superior a 0,05; por lo que no hay diferencia estadísticamente significativa entre las medias de las 2 variables para un 95,0 % de confianza. Para supositorios de 500 mg de dosis para adultos se obtuvo un valor de p de 0,8400, por lo que el comportamiento fue similar.
Teniendo en cuenta los resultados obtenidos se pueden sugerir la aplicación de ambos métodos al control de calidad de supositorios de naproxeno, ya que resultaron válidos con el objetivo con que fueron diseñados.
REFERENCIAS BIBLIOGRÁFICAS
1. Insel PA. Analgésicos-antipiréticos, antiinflamatorios y fármacos antigotosos. En: Goodman LS & Gilman A. Las bases farmacológicas de la terapéutica. 9na ed. Vol. I, Cap. 27. Monterrey: Healtcare Group; 2002. p. 661-705.
2. Feria M. Fármacos analgésicos, antitérmicos y antiinflamatorios no esteroideos. Antiartríticos. En: Florez J. Farmacología humana. 4ta ed. Cap. 22. Barcelona: Editorial: Masson; 2005. p. 393-4.
3. Ansel HC, Popovich NG, Allen LV. Pharmaceutical Dosage Forms and Drug Delivery Systems. 6ta ed. Baltimore: Lippincott Williams & Wilkins; 1999. p. 101-6.
4. Mora CP, Tello ME, Martínez F. Validación de una metodología analítica para la cuantificación de naproxeno en estudios de reparto líquido/líquido mediante espectrofotometría ultravioleta. Re Col Cienc. Quím Farm. 2006;35:81-105.
5. United States Pharmacopoeial Convention XXX and National Formulary 25st. USP XXX. United States Pharmacopoeia Convention. 30 ed. Rockville: Mack Printing; 2007. Versión electrónica.
6. PLM. Diccionario de Especialidades Farmacéuticas. 36ta ed. Bogotá: Thomson Healthcare; 2008. Versión electrónica.
7. Clínica Universidad de Navarra. Antiinflamatorios no esteroideos vía sistémica. naproxeno, 2009. Disponible en: http://www.cun.es/
8. British Pharmacopoeia. Volume I & II Monographs: Medicinal and Pharmaceutical Substances Naproxen. London: The Stationary Office; 2009. Versión electrónica.
9. Ministerio de Salud Pública. Centro para el Desarrollo de la Farmacoepidemiología. Formulario Nacional de Medicamentos. Cap. 2. La Habana: Editorial Ciencias Médicas; 2006. p. 30, 33.
Recibido: 15 de febrero de 2011.
Aprobado: 8 de abril de 2011.
MSc. Yaslenis Rodríguez Hernández. Instituto de Farmacia y Alimentos (IFAL). Universidad de La Habana. Avenida 23 No. 21 425 e/ 214 y 222, La Coronela, La Lisa, La Habana, Cuba. Correo electrónico: yania_as@yahoo.es; yaniasp@ifal.uh.cu


{kind=link}
{kind=link}