










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Medicina
versión On-line ISSN 1561-302X
Rev cubana med v.38 n.4 Ciudad de la Habana oct.-dic. 1999
Temas Actualizados
Instituto de GastroenterologíaMediadores bacterianos de la inflamación en la gastritis crónica por Helicobacter pylori
Dr. Felipe N. Piñol Jiménez1 y Dr. Manuel Paniagua Estévez2RESUMEN
Se conoce que la inflamación crónica del estómago es un tema muy controvertido en la práctica médica, desde los puntos de vista clínico, endoscópico, radiológico e histológico. Se investiga arduamente para revelar los diferentes mecanismos fisiopatológicos que producen el daño hístico del estómago. Con el descubrimiento y caracterización del Helicobacter pylori y su estrecha relación con la gastritis crónica tipo B y las úlceras gastroduodenales, la historia natural de estas afecciones y su abordaje terapéutico han cambiado radicalmente. Se exponen en este trabajo los diferentes procesos que ocurren tras su llegada al estómago, se resaltan las acciones que producen los mediadores bacterianos (proinflamatorios y antigénicos) que participan en el daño hístico, traducido en la clínica como gastritis y que constituye un tema de gran interés para clínicos, gastroenterólogos y médicos en general interesados en la búsqueda de nuevas terapéuticas que no sólo eliminen la bacteria sino que también regulen la acción de los mediadores bacterianos para una reparación adecuada de la mucosa gástrica.Descriptores DeCS: MEDIADORES DE INFLAMACION/fisiología; GASTRITIS/fisiopatología; HELICOBACTER PYLORI.
La inflamación crónica del estómago constituye uno de los temas más controvertidos en la práctica médica desde los puntos de vista clínico, endoscópico, radiológico e histológico. Año tras año se investiga arduamente con el fin de revelar los diferentes mecanismos fisiopatológicos que provocan el daño hístico del estómago.
Numerosas hipótesis tratan de explicar el comienzo y el final del daño hístico provocado en la mucosa gástrica por diferentes agentes, así como sus respectivas complicaciones locales y sistemáticas. Como factores involucrados se destacan los: genéticos, ambientales (infecciosos, dietéticos, consumo de tabaco y drogas), vasculares, psicológicos, inmunológicos, nerviosos, entre otras alteraciones.1,2
Con el descubrimiento y caracterización del Helicobacter pylori y su estrecha relación con la gastritis crónica tipo B y las úlceras gastroduodenales, la historia natural de estas afecciones y su abordaje terapéutico han cambiado radicalmente.3-5
Por la importancia que se le imprime en la actualidad a la infección por Helicobacter pylori y la afirmación indiscutible de que es el agente etiológico más común de la inflamación gástrica, describiremos los diferentes procesos que ocurren tras su llegada al estómago.
La patogénesis de la gastritis crónica por Helicobacter pylori incluye 2 etapas: la primera está caracterizada por la llegada y penetración del microorganismo al mucus gástrico donde se asienta y se multiplica. En esta etapa la bacteria libera varias sustancias tóxicas que son capaces de estimular la respuesta inmunológica local, expresada en un aumento de IgA secretora, con el fin de evitar el proceso de la infección. Las principales células inflamatorias participantes en este proceso inicial son los neutrófilos, que son atraídos al sitio de la lesión; de ahí que su presencia en compañía de folículos linfoides se considere como "signo de actividad". Durante esta fase es frecuente observar la invasión de Helicobacter pylori en las células epiteliales (fig.1).6-8
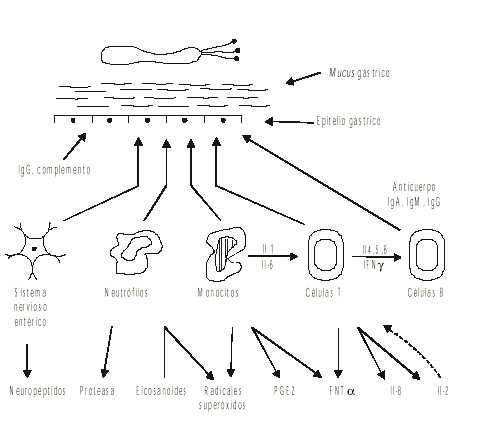
En la segunda etapa se presenta una amplificación de la respuesta inflamatoria, por la interacción de linfocitos, neutrófilos, macrófagos, células mastoides y células no inmunes que, al ser atraídos al sitio de la lesión, liberan gran variedad de mediadores químicos como: citoquinas, eicosanoides, metabolitos reactivos de oxígeno (radicales libres de oxígeno) y el sistema de complemento, que perpetúan la inflamación (fig.2).9-12
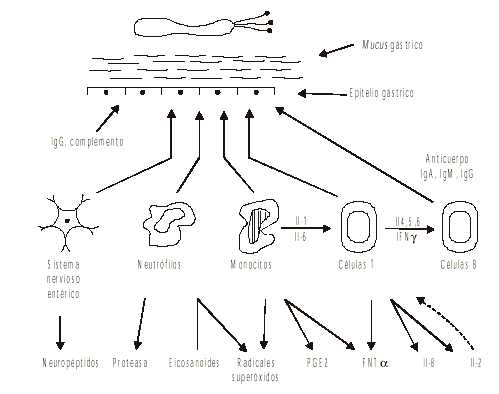
En esta última etapa, también participan los neuropéptidos liberados por las neuronas del sistema nervioso entérico, que contribuyen a ampliar la respuesta inflamatoria y aumentan los daños funcionales del estómago colonizado por Helicobacter pylori.13 La segunda etapa es importante en la patogénesis de la inflamación gástrica; resalta la participación del sistema inmune local y sistémico en el control de la infección y la neutralización de las toxinas bacterianas. Además,se potencializa la destrucción hística que, según su intensidad y duración, puede crear una úlcera gastroduodenal.
Independientemente de los procesos explicados en la patogénesis de la gastritis por Helicobacter pylori, es evidente que la capacidad del germen para inducir respuesta inmune y activación celular varía en las diferentes cepas reconocidas: las tipo I (CagA) poseen mayor capacidad para inducir el proceso inflamatorio en la mucosa gástrica que las tipo II (VacA), que son menos citotóxicas.14,15
Las variaciones en la capacidad del Helicobacter pylori para inducir respuesta inmune, nos han motivado a realizar una revisión sencilla y detallada de los diferentes mecanismos fisiopatológicos desencadenados por la infección de esta bacteria al liberar sus toxinas cuando llega al estómago.
MEDIADORES BACTERIANOS DE LA INFLAMACIÓN
La asociación de la infección por Helicobacter pylori y las afecciones gastroduodenales crónicas es un estímulo constante para miles de investigadores que tratan de esclarecer cómo una bacteria gramnegativa, poco invasora y que habita en la mucosa gástrica es capaz de desarrollar gastritis crónica activa, úlcera gastroduodenal, gastritis atrófica, cáncer gástrico y linfoma gástrico tipo MALT.Como se ha afirmado anteriormente, en el proceso inflamatorio de la mucosa gástrica por Helicobacter pylori participa un grupo de sustancias tóxicas liberadas por la bacteria que desencadenan e inician el daño histológico. Asimismo, esta capacidad de producir toxinas citotóxicas es variable en las distintas cepas de Helicobacter pylori reconocidas, lo que explica la existencia de una gran heterogeneidad genética de Helicobacter pylori para desencadenar el daño hístico.14
Dentro del grupo de mediadores bacterianos de la inflamación se destacan los proinflamatorios1,6,8,15-18 y las sustancias antigénicas.
MEDIADORES BACTERIANOS DE LA INFLAMACIÓN1,6,8,15-18
MediadoresClasificación bacterianos
Proinflamatorios Ureasa
Proteasa
Lipasa y fosfolipasa A2 y C
Superóxido dismutasa
Factor activador de plaqueta
vacuolizantes
UREASA
Una de las características más importantes de las cepas de Helicobacter pylori oxidasa y catalasa positivas, es la presencia de una potente enzima denominada ureasa, la cual protege a la bacteria de los efectos letales del ácido gástrico mediante la formación de una -nube de amonio- que le sirve para tamponizar su entorno vital y poder colonizar el epitelio.La ureasa se localiza en el espacio periplasmático y en la membrana más externa de la bacteria, su actividad se beneficia cuando el pH es bajo. La producción de ureasa interviene en la regulación del metabolismo de la urea, forma dióxido de carbono y amoníaco.6,19
En diversos trabajos se señala la función tóxica del amoníaco sobre las células eucariotas de la mucosa gástrica, aunque algunos autores opinan que el amoníaco en sí no daña la célula sino que el daño es provocado por uno de sus metabolitos (denominado monocloramina), formado por la interacción del amoníaco con el ácido hipocloroso producido por los neutrófilos activados.20,21
El amoníaco producido por la ureasa difunde más fácilmente que el ion amonio, derivado de la unión del amoníaco y el ácido clorhídrico del jugo gástrico, por lo cual el amonio no se considera dañino para la mucosa gástrica.21
En estudios experimentales, tanto con animales como en el hombre, infectados por Helicobacter pylori, la concentración de amoníaco en el jugo gástrico se encuentra elevada en comparación con los no infectados. Murakami y otros reportan que el amoníaco causa daño mucosal porque, al actuar como agente necrotizante, altera el funcionamiento mitocondrial, la respiración celular y el metabolismo energético, con lo cual disminuye la vitalidad de las células y se produce su muerte.22
El amoníaco es capaz de modificar la secreción gástrica al estimular la secreción de gastrina e incrementar la producción de ácido clorhídrico que alteran la barrera mucosa gástrica y con lo cual se favorece la retrodifusión de hidrogeniones y se provoca más daño hístico.23
Otros mecanismos que se involucran en el daño producido por el amoníaco sobre el epitelio gástrico son: inhibición de la liberación del factor estimulador de crecimiento epidérmico, potente efecto inhibidor del ciclo de Krebs, caída significativa de la mucina intercelular y alteraciones de la microcirculación gástrica (estasis), así como disrupción y necrosis de la capa superficial del epitelio.21,23,24
El amoníaco y otras sustancias liberadas por las bacterias son capaces de reducir la actividad bactericida de las células polimorfonucleares y de los monocitos, al inhibir la acidificación de los lisosomas durante la fagocitosis.25
CATALASA
Es una de las enzimas producidas por la bacteria que desempeña una función importante como factor de virulencia, favorece la sobrevivencia de la bacteria en el tejido inflamado, la protege de las acciones fagocíticas de los neutrófilos, de los metabolitos reactivos de oxígeno (MRO) y la de otros mediadores químicos de la inflamación.18,26,27PROTEASA
Enzima que desintegra la estructura polimérica del mucus y debilita su función como barrera por la pérdida gradual de su viscosidad, lo cual aumenta la retrodifusión del ion hidrógeno.28LIPASA Y FOSFOLIPASAS A2 Y C
Son factores de virulencia importantes. Estas sustancias, liberadas por la bacteria en el sitio de la lesión, son capaces de degradar los fosfolípidos del mucus y disminuir su hidrofobicidad, como consecuencia de su fuerte actividad lipolítica, de ahí su importancia en la ulcerogénesis.29La lipasa y las fosfolipasas A2 y C, al generar lisofosfolípidos provistos de actividad lítica, pueden atacar la integridad de la membrana epitelial y favorecer la liberación de ácido araquidónico (AA), con la consiguiente producción de leucotrienos y otros eicosanoides que contribuyen a la inflamación. Estos compuestos, además de su acción inflamatoria, también alteran la permeabilidad de la membrana celular y la regeneración del mucus.30,31
SUPERÓXIDO DISMUTASA
Enzima que se encuentra en altas concentraciones dentro del citoplasma del Helicobacter pylori, utilizada por dicho microorganismo como mecanismo de defensa contra el ataque fagocítico de los neutrófilos. Actúa como antioxidante al catalizar los metabolitos reactivos de oxígeno producidos por los neutrófilos, que pudieran dañarla. Estos hallazgos sugieren que el Helicobacter pylori posee sus propios mecanismos de defensa que contribuyen a su acción patogénica sobre la mucosa gástrica.31FACTOR ACTIVADOR DE PLAQUETAS
La bacteria es capaz de sintetizar y liberar cantidades importantes del factor activador de plaquetas, con potente acción quimiotáctica sobre los neutrófilos y los eosinófilos, así como otras acciones inmunomoduladoras, incluyendo la proliferación de los linfocitos. Este factor es conocido también como un agente proulcerogénico en la mucosa gástrica por su acción sobre la adherencia y activación de los neutrófilos.17,28,31MEDIADORES ANTIGÉNICOS DEL HELICOBACTER PYLORI
Entre los mediadores antigénicos del Helicobacter pylori se encuentran: los lipopolisacáridos, las citotoxinas y las toxinas vacuolizantes que por mecanismos diferentes son capaces de dañar el funcionamiento y el metabolismo energético celular.1,6,8,15 18LIPOPOLISACÁRIDOS
Numerosos trabajos reportan que la membrana externa que cubre la bacteria, es capaz de actuar como material antigénico y estimular la respuesta inflamatoria.La membrana externa de la bacteria es rica en lipopolisacáridos, que son proteínas heterogéneas con baja actividad biológica, capaces de activar los monocitos y los neutrófilos; éstos, a su vez liberan citoquinas, eicosanoides, metabolitos reactivos de oxígeno, activan el complemento en el sitio de la lesión y perpetúan la respuesta inflamatoria, como mecanismo de defensa ante los daños de la bacteria, que al producir más lipopolisacáridos provoca lesión hística local y síntomas sistémicos (fiebre), por lo cual los lipopolisacáridos constituyen uno de los principales antígenos del Helicobacter pylori.32
CITOTOXINAS Y TOXINAS VACUOLIZANTES
La gastritis crónica se asocia con un aumento de la presencia de Helicobacter pylori citotoxigénico, que evoluciona hacia una metaplasia intestinal, lesión considerada como precancerosa en estos casos.La capacidad de producir citotoxinas y toxinas vacuolizantes difiere en los tipos de cepas reconocidos actualmente, lo que permite clasificarlas en 2 grupos: el tipo I, que incluye las cepas con capacidad de secretar proteínas citotóxicas y toxinas vacuolizantes (CagA), de gran importancia en la patogenia de las enfermedades gastroduodenales y el tipo II, que incluye las cepas que no secretan toxinas vacuolizantes ni citotoxinas (VacA), por lo cual tienen una incidencia menor en la inflamación gástrica que las del tipo I.14,33
Las citotoxinas de 82-87 KDa, producidas por Helicobacter pylori, causan vacuolización de las células epiteliales gástricas e inducen una respuesta inflamatoria local, presente en 66 % de la muestra con Helicobacter pylori positivo estudiada por Tummorru y otros.30
Una segunda citotoxina producida por Helicobacter pylori es la proteína de 120- -128 KDa, del grupo de las citotoxinas asociadas al gen A (CagA), que es también un marcador de los efectos vacuolizantes de la bacteria.14
Es interesante plantear que la citotoxina vacuolizante tiene una secuencia molecular no muy diferente de la enzima H-K ATP-ASA que está presente en el canal excretor de las células parietales en el hombre.33 Esto explica la existencia de un grupo de casos de anaclorhidria durante la infección aguda por Helicobacter pylori, por lo cual muchos se preguntan si es a través de este mecanismo que los inhibidores de la bomba de protones por sí mismos pueden actuar contra la bacteria.34
Otro factor importante en la respuesta inflamatoria causada por la bacteria es la adhesión bacteriana, relacionada con la ulcerogénesis y la internalización. La adhesión es considerada como un agravante del daño celular, por este motivo se ha sugerido que la adhesividad tiene mayor relación con el efecto tóxico hístico que con la colonización.9
La adherencia del Helicobacter pylori es específica del tejido gástrico, donde se adhiere al glicocálix y crea puntos de unión que dan lugar a la destrucción de la superficie epitelial y a la pérdida de "microvilli".30,34
En fin, todos estos mediadores bacterianos son capaces de dañar la mucosa y activar el sistema inmune, que libera mediadores químicos capaces de amplificar la respuesta inflamatoria. Estos hallazgos permiten definir a la gastritis asociada a Helicobacter pylori, como una entidad de etiología infecciosa, clínicamente traducida como síndrome dispéptico de tipo no ulceroso de la gastritis.
Fig. 1. Primera etapa del proceso inflamatorio de la mucosa gástrica a la llegada del Helicobacter pylori.
Fig. 2. Segunda etapa del proceso inflamatorio de la mucosa gástrica por Helicobacter pylori. Amplificación de la respuesta inflamatoria por los mediadores químicos liberados por las células del sistema inmune, sistema nervioso estérico y el sistema de complemento.
SUMMARY
It is known that the chronic inflammation of the stomach is a very controversial topic in medical practice from the clinical, endoscopic, radiological and histological points of view. A detailed investigation is being carried out to reveal the different physiopathological mechanisms causing the histic damage of the stomach. With the discovery and characterization of Helicobacter pylori and its close relationship with type B chronic gastritis and gastroduodenal ulcers, the natural history of these affections and their therapeutic approach have changed radically. The different processes that occur after it reaches the stomach are explained in this paper. The actions produced by the bacterial mediators (proinflammatory and antigenic) that take part in the histic damage considered in the clinic as gastritis are also stressed here. This is a topic of great interest for clinicians, gastroenterologists and doctors in general, who are interesed in searching new treatments not only to eradicate the bacteria but also to regulate the action of the bacterial mediators to attain an adequate repair of the gastric mucosa.Subject headings: INFLAMMATION MEDIATORS/physiology; GASTRITIS/physiopathology; HELICOBACTER PYLORI.
REFERENCIAS BIBLIOGRÁFICAS
- Lam SK. Etiology and pathogenesis of peptic ulcer. J Gastroenterol 1994;29(Suppl 7):39-54.
- Crabtree JE. Immune and inflammatory responses to Helicobacter pylori infection. Scand J Gastroenterol 1996;31(Suppl 215):3-10.
- Fax JG. Non-human reservoirs of Helicobacter pylori. Aliment Pharmacol Ther 1995;9:93-103.
- Pignataro S. Helicobacter pylori: reservorios no humanos. Acta Gastroenterol Latinoam 1996;26:34-5.
- Tytgat GNJ, Noach LA, Rauws EAJ. Helicobacter pylori infection and duodenal ulcer disease. Gastroenterol Clin North Am 1993;22:127-39.
- Tytgat GNJ. Helicobacter pylori: recent development. J Gastroenterol 1994;29:30-3.
- Graham DY. Pathogenic mechanisms leading to Helicobacter pylori-induced inflammation. Eur J Gastroenterol Hepatol 1992;4:S9-S16.
- Blaser MJ. Hypothesis on the pathogenesis and natural history of Helicobacter pylori-induced inflammation. Gastroenterology 1992;102:720-7.
- Ishihara S, Fukuda R, Fukumoto S. Cytokine gene expression in the gastric mucosa: Its role in chronic gastritis. J Gastroenterol 1996;31:485-90.
- Fukada T, Kimura S, Arakawa T. Possible role of leukotriens in gastritis associatd with Campylobacter pylori. J Clin Gastroenterol 1990;12:S131-4.
- Nielsen H, Andersen LP. Activation of human phagocyte oxidative metabolism by Helicobacter pylori. Gastroenterology 1992;103:1747-53.
- Dixon MF. Pathophysiology of Helicobacter pylori infection. Scand J Gastroenterol 1994;29 (Suppl 201):7-10.
- Murakami K, Fujioka T, Shiota K, Fujiyoma K, Kodama R, Kawasaki Y, et al. Influence of Helicobacter pylori infection and the effects of its eradication on gastric emptying in non-ulcerative dyspepsia. Eur J Gastroenterol Hepatol 1995;7:S93-7.
- Katz J. ?Cuáles son y qué importancia reviste las distintas cepas patógenas del Helicobacter pylori (H. pylori) en el desarrollo de la enfermedad asociada a la infección? Acta Gastroenterol Latinoam 1996;26:33-4.
- Crabtree JE, Fasmery SM. Helicobacter pylori and gastric mucosal cytokines: evidence that CagA-positive strains are more virulent (editorial, comment) Lab Invest 1995;73:742-5.
- Eaton A, Brooks CL, Morgan DR, Krakowka S. Essential role of urease in pathogenesis of gastritis induced by Helicobacter pylori in gnotobiotic piglets. Infect Immunol 1991;59:2470-5.
- Denizot Y, Sobhani I, Rambaud JC, Lewin M, Thomas Y, Benveniste J. PAF-acether synthesis by Helicobacter pylori. Gut 1990;31:1242-5.
- Hazell SL, Evans DJ, Graham DY. Helicobacter pylori catalase. JGM 1991;137:102-6.
- Ferrero RL, Lee A. The importance of ureasa in acid protection for the gastric-colonising bacteria Helicobacter pylori and Helicobacter felis sp. nov. Microbiol Ecol Health Dis 1991;4:121.
- Visek WJ. Some aspects of ammonia toxicity in animals cells. J Dairy Sci 1968;51:286-95.
- Tsujii M, Kawano S, Tsuji S, Fusamoto H, Kamada T, Sato N. Mechanism of gastric damage induced by ammonia. Gastroenterology 1992;102:1881-8.
- Murakami M, Mizumo M, Ashida Y. NH3-induce gastric lesion, a new model of experimental ulcer. Jpn J Gastroenterol 1986;83:102.
- Levi S, Beardshall K, Ghesan H. Campylobacter pylori and duodenal ulcers: the gastrin link. Lancet 1989;1:1167-8.
- Tsujii M, Kawano S, Tsuji S. Cell kinetics of mucosal atrophy in rat stomatch induced by long-term administration of ammonia. Gastroenterology 1993;104:796-801.
- Klempner MS, Styrt B. Alkalinicing the intralysosomal pH inhibits de granulation of human neutrophili. J Clini Invest 1983;72:1793-1800.
- Hezell SL, Menoz GL. The metabolism and enzymes of Helicobacter pylori: function and potential virulence efects. En: Goodwin CS, Worsley BW, eds. Helicobactr pylori. Biology and clinical practice. Boca Raton: CRC Press, 1993:115-41.
- Doweck J. Respuesta inmune al Helicobacter pylori. Acta Gastroenterol Latinoam 1996;5(Suppl):35-6.
- Slomiany BL, Bilski J, Sarosiek J. Campylobacter pyloridis degrades mucin and undermines gastric mucosal integrity. Biochem Biophys Res Commun 1987;144:307-14.
- Langton SR, Cesareo SD. Helicobacter pylori associated phospholipase A2 activity: a factor in peptic ulcer production? J Clin Pathol 1992;45:221-4.
- Wallace JL. Lipid mediators of inflammation in gastric ulcer. Am J Physiol 1990;(61):258.
- Dobrilla G. Helicobacter pylori. Mecanismo patogénicos. En: Helicobacter pylori. Patogenia. Curso de perfeccionamiento profesional. Aparato Digestivo. Barcelona: Fundación Promedio-Promoción Médica, 1995:11-30.
- Mai U, Pérez-Pérez GI, Wahl LM, Wahl SM, Blaser MJ, Smith PD. Soluble surface proteins frome Helicobacter pylori activate monocytes/macrophages by lipopolysaccharide-independent mechanism. J Clin Invest 1991;87:894-900.
- Tummuru M, Cover TL, Blaser MJ. Cloning and expression of a high-molecular-mass major antigen of Helicobacter pylori: evidence of linkage to cytotoxin production. Infect Immunol 1993;61:1799-1809.
- Tytgat GNJ, Lee A, Graham DY, Dixon MF, Rokkas T. The role of infections agents in peptic ulcer disease. Gastroenterology 1993;6:76-89.
Recibido: 14 de enero de 1999. Aprobado: 29 de abril de 1999.
Dr. Felipe N. Piñol Jiménez. Instituto de Gastroenterología, calle 25 No. 503, entre H e I, El Vedado, Ciudad de La Habana, Cuba.

