










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Medicina
versión On-line ISSN 1561-302X
Rev cubana med v.40 n.4 Ciudad de la Habana oct.-dic. 2001
De la prensa médica extranjera Puesta al día
Genética y biología molecular en cardiología (III) Infección y aterosclerosis
Enrique P. Gurfinkel
Unidad Coronaria. Fundación Favaloro. Buenos Aires. Argentina.
Resumen
Hasta el comienzo del siglo XIX y en el mundo entero, la infección fue la causa dominante de la mortalidad, particularmente la tuberculosis y las diarreas infecciosas. Los avances sanitarios y la introducción de los antibióticos redujeron de forma espectacular la prevalencia de estas entidades. Tras las grandes guerras mundiales, la supervivencia de la humanidad se incrementó de manera significativa, asociada a un dinámico trabajo de investigación científica, particularmente en los estudios de mecanismos fisiopatológicos de los siguientes desafíos médicos: el cáncer y la aterosclerosis. Cerca del final de la frontera entre el conocimiento de estos y la patología, el curso de la investigación giró rápidamente y, una vez más, hacia la infección: el asma, la úlcera péptica y el síndrome de inmunodeficiencia adquirida del adulto, aparecieron fuertemente asociadas a microorganismos. De todos modos, se continuaron publicando descubrimientos sobre receptores hormonales, asociaciones genéticas, incluyendo el reciente desarrollo del proyecto “genoma humano”. Sin embargo, las agencias gubernamentales dedicadas a la salud continuaron hasta el comienzo del siglo xx indicando que, a pesar de todos los avances en las distintas disciplinas, la aterosclerosis, a través de sus clásicas expresiones sintomáticas, seguía siendo la primera causa de muerte de la humanidad. Hacia finales de la década de los años noventa, algunos reducidos ensayos clínicos que utilizaron antibióticos macrólidos en esta población alertaron a la comunidad científica. ¿Habíamos olvidado algo?
Palabras clave: Infección. Aterosclerosis. Inmunidad. Inflamación.Summary
Up to the beggining of the XIX century and in the whole world, infection was the dominant cause of mortality, particularly tuberculosis and infectious diarrheas. The health advances and the introduction of antibiotics reduced the extraordinary way of prevalence of these diseases. After the big world wars, the survival of humanity increased significantly, associated with a dynamic work of scientific research, specially in the study of physiopathological mechanisms of the following medical challenges: cancer and atherosclerosis. Almost at the end of the border between the knowledge of these mechanisms and pathology, the course of research changed fast and, once more, towards infection: asthma, peptic ulcer and acquired immunodeficiency syndrome in the adult appeared strongly associated to microorganisms. Anyway, the publication of the discoveries on hormonal receptors and genetic associations, including the recent development of the “human genoma” project, continued. However, until the begining of the XX century the government health agencies still considered that in spite of the advances achieved in the different disciplines, atherosclerosis, through its classic symptomatic expressions, was the first cause of death for humanity. At the end of the l990s, a few clinical trials that used macrolide antibiotics in this population warned the scientific commmunity. Have we forgotten anything?
Key Words: Infection. Atherosclerosis. Immunity. Inflammation.
Introducción
La enfermedad aterosclerótica y en particular la coronaria, es la entidad nosológica más prevalente en la humanidad y su progresión hacia manifestaciones clínicas severas parece tener una demostrada relación con ciertos factores de riesgo cardiovasculares tradicionales como la diabetes, dislipemias y el hábito tabáquico. Sin embargo, cerca de la mitad de los pacientes que sufren un infarto de miocardio no presentan ningún factor de riesgo convencional identificable.1 Este hecho ha dado un renovado impulso a la investigación de mecanismos alternativos que expliquen la génesis y evolución de las placas ateroscleróticas.
La identificación de la formación trombótica inmediatamente después del accidente de una placa aterosclerótica2 permitió suponer que este estadio final constituido tanto de lisis como de generación de trombina no debería cesar de forma inmediata,3 incluso tras iniciar una terapéutica antitrombótica efectiva,4 lo que explicaría la elevada tasa de recurrencia de acontecimientos isquémicos que se observaba en la práctica clínica. Este conjunto de evidencias coincidieron temporalmente con las observaciones patológicas procedentes de la utilización de nuevas técnicas inmunohistoquímicas,5 las cuales sugirieron un curioso comportamiento local de un fenómeno hasta entonces poco considerado en este contexto: la inflamación.6
Revisaremos a continuación los mecanismos conocidos de acción inflamatoria e inmunológica relacionados con las enfermedades coronarias agudas y crónicas, y su vinculación con los patógenos intracelulares. Nos referiremos, finalmente, al posible papel que pueden tener algunos antibióticos, como los macrólidos, utilizados como complemento terapéutico de las estrategias convencionales.
La carga infecciosa y el sistema inmunológico
El material obtenido por aterectomía direccional de placas ateroscleróticas coronarias en pacientes agudos permitió comprobar la existencia de una elevada concertación de células macrófagas y células inflamatorias activadas en ellas, contrariamente a situaciones clínicas estables con placas sin alteraciones morfológicas.7 El conocimiento acumulado nos permite hoy día formular la siguiente cascada de acontecimientos conducentes a un fenómeno trombótico agudo: un fenómeno inflamatorio activo, probablemente sistémico, se sucede en la pared vascular, lo cual predispone a la vulnerabilidad de una placa determinada a alterarse en su aspecto “topográfico” y dar como resultado la generación de un coágulo firme en trombina que, parcial o totalmente oclusivo, concluirá en un daño miocárdico y su inmediata repercusión en la función de su actividad como bomba mecánica cardíaca. Según esta hipótesis, debió existir previamente un daño sobre el endotelio vascular desencadenado por señales complejas mucho tiempo atrás, entre las células del endotelio, células de músculo liso, monocitos, macrófagos y células T. Esta alteración endotelial, una vez ocurrida, cambiará de forma significativa su naturaleza.
Se supone que el endotelio “intacto” mantiene una acción inhibitoria sobre la adhesión y activación de las células circulantes y es, además, fuente de factores de crecimiento, como de relajación y constricción. Sin embargo, sus propiedades resultan constatemente afectadas por estímulos físicos y químicos, en particular crónicos, que pueden transformarlo “disfuncionante”. El daño del endotelio vascular es aún la teoría más atractiva sobre la génesis de la aterosclerosis, denominada “respuesta a la agresión” inicialmente propuesta por Ross y Glomset en 1976.8 Modificada varias veces, aun sobre las bases iniciales de las ideas de Virchow, incorpora las recientes evidencias de la interacción celular y la inflamación. Así, un estímulo crónico como la infección puede inducir la adhesión y migración de monocitos hacia la íntima, sin necesidad de solución de continuidad alguna en la capa endotelial. Este mecanismo de adhesión probablemente sea facilitado por las condiciones reológicas del flujo, en especial el estrés de rozamiento.9
Por tanto, nos enfrentamos de manera permanente a una innumerable cantidad de microorganismos, algunos patógenos y otros potencialmente definidos como tales que desafían a diario el sistema inmunológico. A lo largo de la evolución, la naturaleza ha desarrollado en el hombre y en otras especies una serie de mecanismos para lograr el estado de inmunidad capaz de perdurar en el tiempo.
El SISTEMA INMUNOLÓGICO
Éste cumple la tarea de detectar la presencia de agentes extraños, de destruirlos de manera específica sin afectar estructuras propias y de recordar su paso por el organismo. Los órganos linfoides primarios y secundarios y una variedad de tipos celulares y moléculas distribuidos por todo el cuerpo integran este sistema. Hay dos tipos de mecanismos de defensa bien diferenciados pero interrelacionados entre sí: mecanismos inespecíficos y específicos.
Los mecanismos inespecíficos, o innatos, son preexistentes, antes de que cualquier agente penetre en el organismo. Se trata de una serie de estructuras primitivas de reconocimiento y destrucción de diversos orígenes. La capacidad de respuesta de las células que intervienen en esta categoría no resulta afectada por el contacto sucesivo con un determinado antígeno. En este sentido podemos decir que no desarrollan “memoria”. En ocasiones, la sola acción de esta rama del sistema inmunológico es suficiente para destruir a un agresor. Contrariamente a éstos, los mecanismos específicos son adquiridos. Se desarrollan frente a la exposición a agentes extraños y poseen la exquisita capacidad de “individualizar” y “memorizar” a cada uno de ellos. Definiremos como antígeno a cualquier molécula que pueda ser reconocida por receptores de linfocitos y, por lo tanto, los active. La región del antígeno que es reconocida por el receptor recibe el nombre de determinante antigénico (DA) o epítope.
Algunos mecanismos inespecíficos de relevancia
Barreras naturales
Las primeras líneas de defensa contra el ingreso de antígenos están constituidas por los epitelios que recubren las superficies externas e internas del organismo. Como ejemplo, la piel indemne constituye una efectiva barrera al paso de los microorganismos, no sólo por su acción física, sino también por la acción química inhibitoria del ácido láctico y de los ácidos grasos de las secreciones sudoríparas y sebáceas.
Fagocitosis
La fagocitosis comprende la ingestión y destrucción posterior de bacterias, células infectadas y partículas inertes por células fagocíticas.
Ésta se inicia con la llegada de las células fagocíticas (macrófagos y neutrófilos polimorfonucleares) al lugar de la agresión, atravesando las paredes de los capilares en respuesta a la acción de sustancias quimiotácticas presentes en la zona. La adhesión del microorganismo a la superficie del neutrófilo o del macrófago provoca la activación de la célula, con lo cual la membrana del fagocito comienza a invaginarse hasta rodear por completo al microorganismo en una vacuola denominada fagosoma. En el citoplasma del fagocito, el fagosoma se fusiona rápidamente con lisosomas para formar un fagolisosoma, donde el microorganismo es expuesto a una serie de sustancias microbicidas provenientes de los lisosomas que terminan, en general, con la lisis del microorganismo ingerido.
Los mecanismos que intervienen en la destrucción del microbio pueden ser dependientes de oxígeno o independientes de él. Los primeros se caracterizan por un notableaumento de la vía de las hexosas fosfato que provoca en última instancia un marcado incremento en el consumo de oxígeno y en la producción de anión superóxido, peróxido de hidrógeno, oxígeno singulete y radical hidroxilo, todos ellos potentes agentes microbicidas. Los mecanismos independientes del oxígeno incluyen la acción de proteínas catiónicas de alto peso molecular, que son capaces de lesionar la membrana microbiana.
Hasta aquí la fagocitosis parece ineludible para un microorganismo que penetre en el cuerpo. Sin embargo, muchos de ellos han adquirido características especiales que les permiten escapar al reconocimiento directo por células fagocíticas. No obstante, la fagocitosis puede llevarse a cabo gracias a la existencia de factores solubles que se unen a la superficie de estos microorganismos evasores y advierten a los fagocitos de la presencia de un elemento extraño. Estos factores son los anticuerpos (Ac) IgG e IgM y la fracción C3b del componente C3 del sistema de complemento. Un microorganismo recubierto por estas moléculas puede ser fácilmente reconocido y captado por macrófagos y polimorfonucleares a través de receptores específicos para el fragmento Fc de las IgG y para C3b presentes en la membrana celular de los fagocitos. Las moléculas capaces de favorecer la fagocitosis en esta forma, como IgG y C3b, se denominan opsoninas y el fenómeno en sí se denomina opsonización. Se ha comprobado que la IgG por sí sola es capaz de mediar la adhesión e ingestión de la célula opsonizada a través de sus receptores específicos en fagocitos, mientras que la opsonización únicamente por C3b sólo promueve la adhesión.
Interferones
Los interferones (IFN) comprenden un grupo de proteínas responsables de mediarel fenómeno de interferencia viral, por el cual un animal infectado con un virus es a menudo resistente a la sobreinfección por un virus diferente. La acción de los interferones ocurre a través de la inducción de un estado antiviral en sus células diana y no por interacción directa con el virus mismo. Además de la actividad antiviral tienen efecto sobre el crecimiento y proliferación de células normales y tumorales y participan en la regulación de la respuesta inmunológica.
Citotoxicidad natural killer
Este fenómeno está representado por la capacidad innata que poseen ciertas subpoblaciones linfocitarias de destruir células tumorales y células infectadas por virus mediante la liberación de sustancias tóxicas al medio extracelular. Las células efectoras se denominan células natural killer o células NK. Al igual que el resto de los mecanismos de defensa inespecíficos, la intervención de las células NK no requieren exposición previa al antígeno para desarrrollarse ni poseen memoria tras la inmunización.
Mecanismos específicos
Las células y moléculas que intervienen en estos procesos son las que integran la rama adaptativa del sistema inmu-nológico. Las células predominantes de este sistema inmunológico son los linfocitos.
Históricamente se han distinguido dos formas de respuesta inmunitaria. Esta distinción resulta de la existencia de dos subpoblaciones linfocitarias diferentes, los linfocitos B y T. La activación de linfocitos B seguida de su diferenciación a células plasmáticas secretoras de anticuerpos es responsable de la inmunidad humoral. Loslinfocitos T no son capaces de secretar anticuerpos tras su activación. En cambio, cumplen una serie de funciones reguladoras y efectoras que determinan lo que se denomina inmunidad celular.
En conjunto, ambas respuestas se caracterizan por la capacidad adaptativa para responder de diferente forma a los distintos antígenos, por la especificidad que presentan y por presentar el fenómeno de memoria inmunológica.
Ambos tipos de células son capaces de reconocer sustancias extrañas a través de receptores presentes en la membrana celular. Es necesaria la presencia de varios tipos celulares en forma simultánea para recrear una respuesta inmunitaria plena, perfectamente regulada. Por ejemplo, los linfocitos B, si bien son capaces de reconocer los distintos antígenos de forma independiente, requieren de ciertas señales accesorias que son aportadas por una subpoblación de linfocitos T denominados linfocitos T helper o colaboradores, para diferenciarse a células plasmáticas productoras de anticuerpos.
Lipoproteínas de baja densidad y el endotelio vascular “disfuncionante”
De forma simultánea a los fenómenos de estimulación crónica a que es expuesto el endotelio vascular, las lipoproteínas de baja densidad (LDL) que circulan en el torrente sanguíneo pueden precipitar en esta misma pared una vez reconocidas por receptores endoteliales. Tras esto, y cuando aún se encuentran en el fluido extracelular, si ingresan en un proceso de oxidación ori ginado por ciertos estímulos no aún totalmente esclarecidos, contribuirán significa-tivamente al fenómeno aterogénico. En caso que no sean oxidadas, podrán volver al torrente sanguíneo sin contribuir en apariencia a este último proceso (fig. 1). La alteración oxidativa provoca, como consecuencia, severas alteraciones en las funciones endoteliales. Por ejemplo, éste pierde su naturaleza vasodilatadora al excitar, entre otras, la enzima sintetasa iNOS (enzima sintetasa inespecífica del óxido nítrico), promoviéndose la vasoconstricción y, adicionalmente, una mayor oxidación intravascular de moléculas de LDL. En efecto, el NO sintetizado por la isoforma i permite su unión con aniones superóxidos, activando los factores de transcripción como el factor nuclear kappa-B (NF-kB), el cual promueve la expresión de moléculas de adhesión celulares como VCAM-1 e ICAM-1, y factores quimioatractivos de monocitos (MCP), con lo que se establece el puente necesario para la adhesión y migración de células inflamatorias.10 Las primeras en activarse reaccionan en cadena activando otras, como macrófagos capaces de generar a su vez numerosos compuestos activos y, además, cambiar la morfología y composición relativa de las placas ateros-cleróticas. Entre los factores más importantes se cuentan las interleucinas, los aniones superóxido, los factores de crecimiento como el factor transformador de crecimiento (TGF b) y el factor derivado de plaquetas (PDGF).

Fig. 1. Desplazamiento de la lipoproteína de baja densidad (LDL) desde el torrente vascular y hacia la pared arterial, así como su mecanismo inverso.
De la placa estable a la inestable por el camino de la información retrospectiva
La presencia de las células inflamatorias es crucial para cambiar una lesión aterosclerótica estable a una placa vulnerable, aunque debemos reconocer que toda esta información es obtenida de forma retrospectiva, una vez ocurridos los hechos. Hasta el momento actual no existe ninguna técnica ni un modelo animal en el cual se haya conseguido producir una placa vulnerable que pueda ser analizada momento a momento hasta su alteración. Por tanto, se presupone que los macrófagos y los linfocitos son las células críticas de la placa aterosclerótica, las cuales van desplazando la concentración de células musculares lisas en la medida en que la placa evoluciona, células anteriormente consideradas como claves de este fenómeno.11 Las células inflamatorias se organizan de forma que la estirpe CD4+ se presenta con más frecuencia en las placas maduras, mientras que las células CD8+ en lesiones ateroscleróticas más jóvenes. Las células CD4+ que se encuentran normalmente junto a los macrófagos, y bajo circunstancias poco establecidas, expresan en su superficie moléculas del sistema mayor de histocompa-tibilidad humana (HLA clase II [DR]) que sugieren una significativa activación celular. Además, una subpoblación celular proveniente de estos CD4+, denominados CD4+CD28+, han sido recientemente hallados en este tipo de pacientes con placas de características inestables, altamente secretoras de interferón gamma,12 aunque este tipo celular ya había sido identificado en la década de los ochenta por otros grupos de investigación sobre reumatología en pacientes con formas severas de artritis reumatoide. Más allá, los macrófagos, gracias a este tipo de señal de activación, se convierten en células que presentan subsecuentemente antígenos a los linfocitos a través de las moléculas del sistema HLA.13
Este ciclo inflamatorio resulta estimulado gracias a la adicional e intensa liberación de citocinas, como la interleucina 1 (IL-1), interleucina 6 (IL-6) y el interferón-g, así como el factor de necrosis tumoral alfa (TNF-a), el cual incrementa adicionalmente la secreción del factor tisular (TF),14 el eslabón inicial de la excitación de la vía extrínseca de la coagulación sanguínea. Este último no sólo parece ser localmente sintetizado, sino también en las células apoptóticas que conviven en la placa aterosclerótica y los monocitos circulantes, lo cual convierte al cuerpo entero en una fuente de trombosis, marcando un estado de activación generalizado para un eventual accidente vascular (fig. 2).
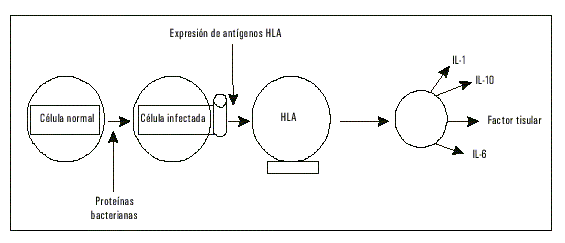
Fig. 2. Interacción celular dentro de la pared vascular con la expresión de antígenos del sistema HLA, clase II (DR).
El grado de complejidad de la actividad inmunológica es tal que el proceso tiene características de afección sistémica. En efecto, algunos marcadores de esta actividad se pueden detectar en la sangre venosa donde drena la arteria comprometida por un proceso trombótico agudo, como también desde venas donde ésta no drena directamente.15
El papel de la carga infecciosa y el sistema inmunológico
El primer agente infeccioso identificado de manera directa con la aterosclerosis fue un tipo apícola de herpesvirus (asociado a la enfermedad de Mareck), el cual fue inoculado en gallinas libres de todo tipo de infección y alimentadas con una dieta rica en colesterol por Fabricant, et al.16 en 1978. Luego del sacrificio de estos animales se apreciaron lesiones ateroscleróticas claramente visibles y de avanzado tipo, similares a las humanas. El mismo grupo inmunizó con posterioridad a este tipo de animales con el fin de prevenir el desarrollo de aterosclerosis tras de la inoculación, con resultados altentadores,17 documentando una particular acumulación de ésteres de colesterol en las células musculares lisas.18
En los siguientes 15 años, la investigación giró más hacia un vínculo intermedio entre la infección y la aterosclerosis dado por el proceso inmunológico, más que por una acción directa de dichos agentes exógenos. Éstos parecen, hoy por hoy, servir como gatillos o mediadores de la activación del proceso inmunitario en la manera en que fue descrita previamente, contribuyendo a la génesis de la placa aterosclerótica, a su cambio de configuración de estable a inestable e incluso a la erosión de la superficie endotelial. Si bien existen datos experimentales importantes relacionados con cada una de estas especies fisiopatológicas, en ningún caso las evidencias son concluyentes.
Los patógenos que han sido vinculados con distintos grados de solidez científica19-22 a la enfermedad aterosclerótica son los herpesvirus, citomegalovirus, Micoplasma pneumoniae, Helicobacter pylori y Chlamydia pneumoniae.
El interés por su posible papel es tal que en los últimos años se han realizado varios estudios clínicos de intervención23 y observación,24 con antibióticos macrólidos en pacientes con enfermedad coronaria, buscando establecer la relación particularmente con C. pneumoniae, un patógeno intracelular.
Este tipo de Chlamydia fue originariamente identificado en 1986 por Grayston.25 Se denominó inicialmente como TWAR (TW-183 y AR-39) en un niño de origen taiwanés que padecía tracoma. Ésta es la segunda bacteria, más prevalente en las neumonías atípicas de la comunidad, cuyo contacto inicial suele ocurrir entre los 5-14 años de edad. La reinfección por C. Pneumoniae a lo largo de la vida es un hecho más que común, hasta el punto de estimarse que el 86 % de la población adulta ha estado o está en contacto con ella.26
Esta especie adquiere dos formas de desarrollo.Un cuerpo elemental extracelular que se añade a la membrana celular emulando una actitud parasitaria, siendo luego incorporado por fagocitosis. Uno de los rasgos más sobresalientes de esta bacteria es la capacidad de incorporarse a las células monocitarias. Ya en el citoplasma se convierte en un cuerpo intracelular capaz de iniciar una fusión binaria y transformarse en un cuerpo de inclusión. Una vez que la célula que ocupa es destruida, continúa su ciclo en las nuevas células que habrá de infectar.
C. pneumoniae ha sido hallado, utilizando diferentes técnicas, en las placas ateroscleróticas humanas con diferentes estadios evolutivos. Estos hallazgos alcanzaron a ser reproducidos en modelos animales. Muhlestein, et al.27 indujeron aterogénesis por medio de la administración de C. pneumoniae en forma inhalatoria. Sin embargo, los agentes bacterianos y virales comparten características que hacen difícil confirmar la relación de causa-efecto con la aterosclerosis e inestabilidad de la placa, particularmente debido a los métodos utilizados de diagnóstico, la alta prevalencia en la población adulta de títulos de anticuerpos contra estos agentes, dificultades para el cultivo, la existencia de infecciones cruzadas y la elevada tasa de uso de antibióticos por otras enfermedades.
Independientemente de ello, se han intentado esbozar probables mecanismos fisiopatológicos entre aterosclerosis e infección. Se ha probado que la carga viral induce la secreción de compuestos protrombóticos como el factor tisular28 y la trombina.27 Además, las endotoxinas-estimulan la isoforma inducible del óxido nítrico endotelial (iNOS), como hemos explicado con anterioridad con sus adicionales consecuencias (fig.3).
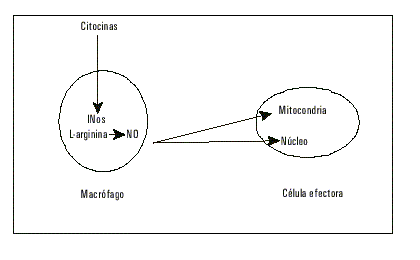
Fig. 3. Consecuencias de la síntesis de óxido nítrico por vía de la enzima sintetasa inducible de óxido nítrico.
Esta carga viral facilita la inducción en el organismo de estímulos de proliferación celular que se ejemplifican en el caso de citomegalovirus, el cual interactúa con la proteína p53, interfiriendo éste con el ciclo celular normal hasta facilitar la reproducción, particularmente de miocitos.29
Varias son las hipótesis que se han formulado para explicar los posibles mecanismos de la relación de H. pylori con la aterosclerosis. Es de especial interés la posibilidad que las infecciones gastrointestinales crónicas produzcan malabsorción y déficit de vitamina B6 y B12 factores esenciales para la conversión de homocisteína en metionina. Por ende, la acción de este tipo de patógenos sería indirecta, vinculada con una hiperconcentración de homocisteína.30 Por otra parte, esta bacteria, al igual que C. pneumoniae, es capaz de sintetizar un antígeno de superficie conocido por su aparición bajo condiciones de inestabilidad térmica, denominado heat shock protein similar a la forma humana 60 (HSP60). Esta familia de proteínas (cercana a una decena) se expresan en el citoplasma cuando existen condiciones como las que dieron su nombre, considerando que normalmente no se observan. Una vez expuestas, pueden no ser reconocidas como propias por el sistema inmunológico del huésped, iniciando así una significativa producción de anticuerpo. Una de ellas, una proteína de 60 KD (HSP60) posee una enorme homología con otra de 65 KD de la bacteria C. pneumoniae (HSP65). Por tanto, un anticuerpo (generalmente ciego en términos de reconocer o diferenciar estructuras) contra ambas proteínas puede iniciar el mecanismo que concluye con daño celular y lisis macrofágica, mediado por una reacción inmunitaria inmediata.31 En apariencia, estetipo de fenómenos parecen vinculados al tipo de antígenos HLA en algunos sujetos32 y esta susceptibilidad explicaría por qué algunos pacientes responden a terapias farmacológicas con independencia de las concentraciones serológicas de anticuerpos anti-Chlamydia33,34 (fig.4).
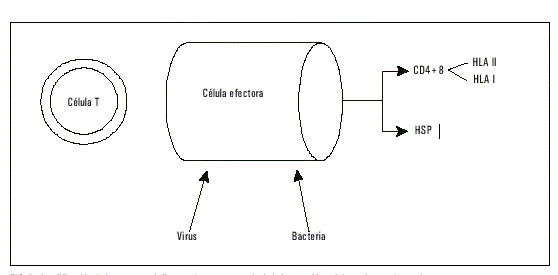
Fig. 4. Amplificación de la respuesta inflamatoria, consecuencia de la interacción celular en la pared vascular.
Estos mismos anticuerpos para HSP 60 guardan relación con la enfermedad difusaaterosclerótica. La exposición de estas proteínas antigénicas contribuye también a estimular la liberación de interleucinas, TNF-a e interferón-g, los cuales vuelven a cerrar viciosamente el círculo inflamatorio.35
Estas evidencias sugieren fuertemente la presencia de un mecanismo auto-inmune, recientemente reproducido gracias a un modelo animal en el que una secuencia peptídica en la pared bacteriana análoga a una de las cadenas aminoacídicas de parte de la miosina pesada pudo provocar una respuesta cruzada autoinmune (fig. 5).36
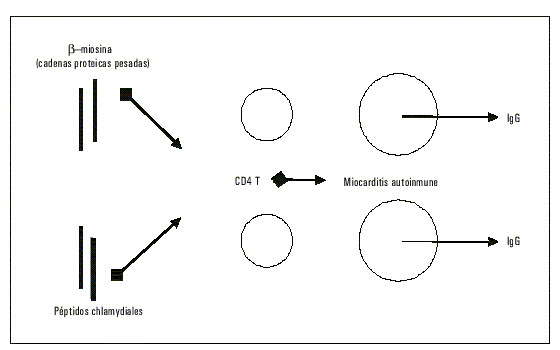
Fig. 5. Respuesta inmunológica en un modelo animal ante la expresión de antígenos y sus epítopes conocidos al sistema inmunológico. El panel inferior indica una respuesta similar cuando antígenos similares son presentados ante iguales células.
Este tipo de infecciones crónicas, en las que los episodios de reinfección son extremadamente frecuentes en la vida, estimulan una y otra vez a un sistema activado con anterioridad, conduciendo a una intensa actividad inflamatoria capaz, entonces, de modificar la composición de la placa.
¿Existe un lugar para los macrólidos en el tratamiento de la enfermedad coronaria?
La presencia de estructuras reconocibles como C. pneumoniae en las placas ateroscleróticas guarda relación proporcional con la severidad de las lesiones,37 lo cual es por completo independiente de las concentraciones serológicas de anticuerpos contra C. penumoniae. Esta interesante observación, así como las dificultades para reproducir en modelos experimentales la secuencia temporal de la formación de las placas ateroscleróticas, abrió paso a una serie de estudios clínicos que investigaron el efecto del tratamiento con macrólidos en pacientes con enfermedad coronaria aguda y crónica.38
Se han publicado hasta el momento tres estudios diseñados para investigar el efecto de los macrólidos sobre la evolución de pacientes con enfermedad coronaria sintomática. Gupta et al.39 incluyeron a 200 pacientes supervivientes de un infarto agudo de miocardio (IAM) a los cuales midieron las concentraciones serológicas de IgG para C. pneumoniae, seleccionando a los 80 que presentaron títulos positivos de manera persistente para un tratamiento aleatorio con 500 mg de azitromicina p.o. durante 3 días, o placebo. Los puntos finales primarios fueron serológicos a 6 meses, etapa en la cual se observó un descenso de los títulos de anticuerpos IgG a < 1: 16 en el 43 % de los pacientes agrupados en el grupo activo frente al 10 % en el grupo placebo (p = 0,02). El análisis secundario puso de manifiesto un aumento de acontecimientos isquémicos mayores en los pacientes tratados con placebo.
Basándose en estas observaciones, Anderson et al.40 diseñaron el estudio Azithromycin in Coronary Artery Disease: Elimination of Myocardial Infection-with Chlamydia, (ACADEMIC), el cual consideró una población de pacientes coronarios estables, de bajo riesgo, tratados con azitromicina p.o. en dosis semanales durante 3 meses, evaluando los puntos finales IgG, TF, citocinas y proteína C reactiva. Los resultados finales no demostraron diferencias en las tasas de acontecimientos clínicos mayores. Teniendo en cuenta el tipo de población, sumado al reducido tamaño de la muestra, era poco probable que se encontraran diferencias en el seguimiento preestablecido a 2 años.41
Este estudio es ciertamente confuso. Los valores plasmáticos de las citocinas proinflamatorias decrecieron a los 90 días, a pesar que el 11 % de la población investigada experimentó nuevas infecciones clínicas intercurrentes, que obviamente debieron afectar a las concentraciones plasmáticas de citocinas proinflamatorias.
El estudio Roxithromycin in Ischemic Síndromes (ROXIS)42 es el único que, por el momento, ha incluido pacientes con síndromes agudos tipo no Q. Éstos fueron asignados de forma aleatoria y doble ciego para recibir por vía oral 300 mg/día de roxitromicina, o su correspondiente placebo, durante 30 días y con independencia de las concentraciones serológicas de anticuerpos anti-C. pneumoniae, por las razones ampliamente descritas con anterioridad, los cuales no suponen de forma alguna una herramienta de identificación de pacientes debido a la amplia divergencia en la bibliografía sobre la existencia de la bacteria en la propia placa, la susceptibilidad del huésped a desencadenar reacciones inmuno-lógicas43 y la dificultad de las técnicas de identificación. Los 202 pacientes incorporados al ensayo recibieron tratamiento convencional completo, incluyendo anticoagulación. Al cabo de 30 días se observó una significativa reducción de acontecimientos primarios en el grupo roxitromicina (muerte, infarto no fatal o isquemia recurrente severa). El análisis secundario practicado durante el segumiento demostró que este beneficio se sostuvo hasta los 90 días, conservando una tendencia favorable para el grupo tratado, aunque no significativa a los 6 meses (fig. 6).
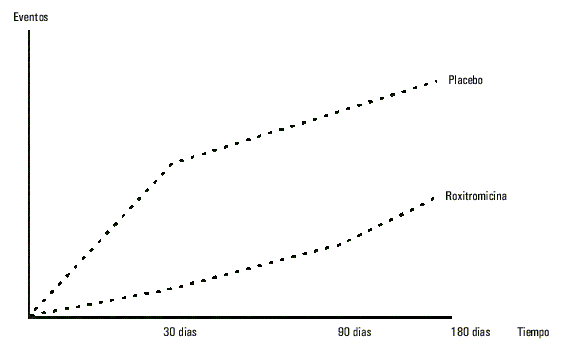
Fig. 6. Acontecimientos clínicos combinados en el estudio Roxis (muerte, infarto no fatal o isquemia recurrente severa). Se observa una reducción significativa a favor del grupo que recibía antibiótico a 30 días. A los 6 meses persiste una tendencia que no alcanza significación estadística.
La medicación fue bien tolerada durante los 30 días iniciales. Los títulos de IgG fueron positivos en aproximadamente la mitad de los pacientes en el momento del ingreso y no variaron después del tratamiento. El número de enfermos con valores elevados de proteína C reactiva, en cambio, fue significativamente menor en el grupo que recibió antibiótico al cabo de un mes, lo cual indica una posible acción antiinflamatoria inespecífica44 (tabla 1).
Tabla 1. Estudios clínicos controlados con administración a macrólidos a pacientes con enfermedad coronaria sintomática
Supervivientes| Estudio y referencia bibliográfica | Antibiótico | Población | Puntos finales, grupo activo | Títulos de IgG/IgA C. pneumoniae | Marcadores de inflamación (PCR, FT, IL-6) |
| ROXIS42 | Roxitromicina | AI/IAM-nQ | ¯ | NS | ¯ |
| Gupta et al.39 | Azitromicina | ¯ | ¯ | ¯ | |
| ACADEMIC40 | Azitromicina | Supervivientes de acontecimientos agudos | NS | NS | ¯ |
NS: No significativo; IAM: Infarto agudo de miocardio; AI: Angina inestable; IAM-nQ: Infarto agudo de miocardio tipo no Q; PCR: proteína C reactiva; FT: Factor tisular.
Selección de pacientes
Como hemos visto, existen estimulantes datos serológicos45 y patológicos46 que indican la relación existente entre patógenos intracelulares, fundamentalmente C. pneumoniae, y la enfermedad aterosclerótica aguda y crónica. No obstante, los datos experimentales no indican aún con claridad que la hora de administrar un tratamiento antibiótico concomitante haya llegado.47 Un análisis de los informes publicados sugiere importantes mensajes para comprender por qué necesitamos más datos experimentales antes de llegar a una conclusión sólida: en primer lugar, los estudios de intervención ha sido hasta el momento exploratorios, por lo que pagan el tributo de un tamaño de muestra relativamente reducido.41
Por otro lado, es clave la selección de pacientes. Tanto en el estudio inglés como en ACADEMIC se incluyeron pacientes supervivientes de infarto de miocardio, cuyas tasas de acontecimientos decrecen con el tiempo. La inclusión de pacientes se basó, además, en criterios serológicos de IgG, lo cual expone al sesgo por las limitaciones explicadas del método. En efecto, la tasa de pacientes seropositivos para C. pneumoniae según la IgG, incluyendo las poblaciones de alto riesgo, una con elevada prevalencia de la bacteria en las placas ateroscleróticas es de un 50-65 %. Los datos recientemente publicados indican que los títulos de IgG e IgA son similares en sujetos con aterosclerosis severa y leve, donde en ambas circunstancias se consigue detectar los anticuerpos anti-C. pneumoniae por inmunofluorescencia directa en el 86 y el 6 % de las placas con estos grados extremos de afectación, respectivamente.
Por último, la descolonización de las arterias puede requerir terapias mucho más prolongadas que las que se han utilizado hasta ahora. Por lo demás, la elevada prevalencia de las colonizaciones expone a las reinfecciones, que es uno de los principales mecanismos que pueden explicar la falta de diferencia en los puntos finales clínicos más allá del día 90, como ha ocurrido durante el estudio ROXIS.
Conclusión
Existe una cadena de procesos inmunológicos e inflamatorios que condicionan el desarrollo, la transformación y la evolución de las placas ateroscleróticas coronarias. Se han identificado patógenos intracelulares, en especial C. pneumoniae, que están relacionados con la severidad de las lesiones y, según los modelos experimentales animales, probablemente con la inducción de la aterosclerosis.48 La elevada prevalencia de colonizaciones crónicas por C. pneumoniae en humanos, así como las limitaciones de la sensibilidad y especificidad de las técnicas de diagnóstico serológicas, hicieron que se avanzara a modelos de estudio piloto en humanos con enfermedad coronaria sintomática. Los resultados del estudio ROXIS en portadores de angina inestable tratados con roxitromicina, y del trabajo de Gupta et al. con azitromicina en supervivientes de infarto de miocardio son alentadores, pero no concluyentes.49
Se han puesto en marcha estudios que han tomado las bases de algunos de los trabajos publicados, que evalúan a gran escala el impacto de terapias prolongadas con azitromicina. Entre ellos, los estudios WIZARD y ACES. Se están sumando a éstos otros ensayos que utilizan quetólidos, macrólidos y quinolonas, los cuales se encuentran próximos a iniciarse y algunos cerca de la finalización de su fase inicial de ingreso de pacientes, por lo que podrán aportar en poco tiempo nuevos indicios sobre uno de los mayores interrogantes surgidos en la cardiología contemporánea.
Bibliografía
- Braunwald E. Shattuck Lecture- Cardiovascular medicine at the turn of the millennium: Triumphs, concerns, and oppurtunities. N Engl J Med 1997;337:1360-1369.
- Braunwald E, Fuster V. Unstable angina. Definition, pathogenesis, and classification. En: Fuster V, Toss R, Topol EJ, editores. Atherosclerosis and coronary artery disease. Filadelfia: JB Lippincot, 1996;1285-1298.
- Gurfinkel E, Bozovich G, Mejaíl I, Cerdá M, Oxilia A, Mautner B. Time significance of acute thrombotic reactant markers in patients with and without silent myocardial ischemia and overt unstable angina. Am J Cardiol 1995;76:121-124.
- Cohen M, Demers C, Gurfinkel E, Turpie AGG, Fromell GJ, Goodman S et al. Eficacy and safety of enoxaparin in non Q wave coronary syndromes. The ESSENCE Study. N Engl J Med 1997;337:447-452.
- Van der Wal AC, Becker A, van der Loos CM, Das PK. Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation 1994;90:1662-1668.
- Ross R. Atherosclerosis - an inflammatory disease. N Engl J Med 1999;340:115-126.
- Galis ZS, Suhkova GK, Lark MW, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest 1994;94:2493-2503.
- Ross R, Glomset JA. The pathogenesis of atherosclerosis. N Engl J Med 1976;295: 369-377.
- Dirksen MT, Van der Wal AC, Van den Berg FM, Van der Loos CM, Becker AE. Distribution of inflammatory cells in atherosclerotic plaques relates to the direction of flow. Circulation 1998;98:2000-2003.
- Cooke PJ. The role of endothelium in atherosclerosis. En: Schultheiss H, Schwimmbeck P, editores. The role of immune mechanisms in cardiovascular disease. Berlín: Springer, 1997; 194-204.
- Moreno PR, Fallon JT. Inflammation in acute coronary syndromes. En: Schultheiss H, Schwimmbeck P, editores: The role of immune mechanisms in cardiovascular disease. Berlín: Springer, 1997;213-29.
- Liuzzo G, Goronzy JJ, Yang H, Kópecky SL, Holmes DR, Frye RL et al. Monoclonal T-cell proliferation and plaque instability in acute coronary syndromes. Circulation 2000;102:2883-2888.
- van der Wal AC, Becker AE, van der Loos CM, Das PK. Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of dominant plaque morphology. Circulation 1994;89:36-44.
- Tannenbaum SH, Finko R, Cines DB. Antibody and immune complexes induce tissue factor production by human endothelial cells. J Immunol 1986; 137:1532-1537.
- Buffon A, Liuzzo G, Donofrio G, Biassuci LM, Maseri A. Intracardiac activation of neutrophils in unstable angina patients in unrelated to culprit lesion. Circulation 1996;94 (Supl):514.
- Fabricant CG, Fabricant J, Litrenta MM, Minick CR. Virus-induced atherosclerosis. J Exp Med 1978; 148:335-340.
- Fabricant CG, Hajjar DP, Minnick CR, Fabricant J. Herpesvirus infection enhances cholesterol and cholesteryl ester accumulation in cultured arterial smooth muscle cells. Am J Path 1981;105:176-184.
- Hajjar DP, Fabricant CG, Minick CR, Fabricant J. Virus-induced atherosclerosis. Herpesvirus infection alters aortic cholesterol metabolism and accumulation. Am J Path 1986;122:62-70.
- Nieto FJ, Adam E, Sorlie P, Farzadeegan H, Melnick JL, Comstock GW et al. Cohort study of cytomegalovirus infection as a risk factor for carotid intimal-medial thickening, a measure of subclinical atherosclerosis. Circulation 1996;94:922-927.
- Gurfinkel EP, Rozlosnik J, Bozovich G, Duronto E, Dos Santos A, Mautner B. IgG antibodies to Chamydia and Mycoplasma infection plus C reactive protein related to poor outcome in unstable angina. Archivos del Instituto de Cardiología de México 1997;67:462-468.
- Benditt EP, Barrett T, McDougall JK. Viruses in the etiology of atherosclerosis. Proc Natl Acad Sci USA 1983;80:6386-6389.
- Murray LM, Bamford KB, O´Reilly DPJ, McCrum EE, Evans AE. Helicobacter pylori infection: relation with cardiovascular risk factors, ischeamic heart disease, and social class. Br Heart J 1995;74:497-501.
- Gurfinkel E. Open and controlled intervention trials for anusual antichlamydia indications: asthma, atherosclerosis, myocardial infarction. En: Allegra L, Blasi F, editores. Chlamydia pneumoniae. Milán, Italia: Springer-Verlag, 1999;185-187.
- Anestad G, Scheel O, Hungnes O. Chronic infection and coronary artery disease (carta). Lancet 1997;350:1028.
- Grayston JT, Kuo C, Wang S, Altman J. A new Chlamydia psittacii strain, TWAR, isolated in acute respiratory tract infections. N Engl J Med 1986;315:161-168.
- Aldous MB, Grayston JT, Wang A, Foy H. Seroepidemiology of Chlamydia pneumoniae TWAR in Seattle Families, 1966-79. J Infect Dis 1992;166:646-649.
- Muhlestein JB, A nderson JL, Hammond EH, Zau L, Trehan S, Schwobe EP et al. Infection with Chlamydia pneumoniae accelerates the development of atherosclerosis and treatment with azythromycin prevents it in a rabbit model. Circulation 1998;97:633-636.
- Epstein SE, Speir E, Zhou YF, Guetta E, Leon M, Finkel T. The role of infection in restenosis and atherosclerosis: focus on cytomegalovirus. Lancet 1996;348:S13-S16.
- Speir E, Modali R, Huang ES. Potential role of human cytomegalovirus and p53 interaction in coronary restenosis. Science 1994;265:391-394.
- Sung JJ, Sanderson JE. Hyperhomocysteinaemia, Helicobacter pylori, and coronary heart disease. Heart 1996;76:305-307.
- Metzler B, Schett G, Kleindienst R, Van der Zee R, Ottenhoff T, Hajeer A et al. Epitope specificity of anti- heat shock protein 65/60 serum antibodies in atherosclerosis. Arterioscler Thromb Vasc Biol 1997;17:536-541.
- Gurfinkel E. Link between intracellular pathogens and cardiovascular disease. Clin Microbiol Infect 1998;4:533-36.
- Jones RB. Chlamydia trachomatis. En: Mandel GL, Bennet JE, Dolin R, editores. The principles and practice of infectious diseases. Nueva York:Churchill Livingstone, 1995;1679-1693.
- Gurfinkel E, Bozovich G. Chlamydia pneumoniae, inflammation and the instability of the atherosclerotic plaque. Atherosclerosis 1998;140(Supl 1):31-35.
- Leinone M. Pathogenetic mechanisms and epidemiology of Chlamydia pneumoniae. Eur Heart J 1993;14(Supl K):57-61.
- Buchmaier K, Neu N, De la Mata LM, Pal S, Hessel A, Penninger JM. Chlamydia infections and heart disease linked through antigenic mimicry. Science 1999;2838:1335-1339.
- Ericson K, Saldeen TGP, Lindquist O, Pahlson C, Mehta JL. Relationship of Chlamydia pneumoniae infection to severity of human coronary atherosclerosis. Circulation 2000;101:2568-2571.
- Gurfinkel E, Bozovich G. Emerging role of antibiotic in atherosclerosis. Am Heart J 1999;138:S537-S538.
- Gupta S, Leatham EW, Carrington D, Mendal MA, Kaski JC, Camm J. Elevated Chlamydia pneumoniae antibodies, cardiovascular events and azithromycin in male survivors of myocardial infarction. Circulation 1997;96:404-407.
- Anderson JL, Muhlestein JB, Carlquist J, Allen A, Trehan S, Nielson C et al. Randomized secondary prevention trial of azythroycin in patients with coronary artery disease and serological evidence for Chlamydia pneumoniae infection. Circulation 1999;99:1540-1547.
- Gurfinkel E, Bozovich G, Mautner B. Chlamydia pneumoniae in coronary artery disease [carta]. Circulation 2000 14;101:E95.
- Gurfinkel E, Bozovich G, Daroca A, Beck E, Mautner B. Randomized trial of roxithromycin in non-Q-wave coronary syndromes: ROXIS pilot study. Lancet 1997;350:404-407.
- Raimondi E, Gurfinkel E, Bozovich G, Padrós K, Rodríguez B, Berardi G etal. Coronary artery disease in Argentina. En: Gjertson DW, terasaki PJ, editores. HLA Nueva York: American Society for Histocompatibility and Immunogenetic, 1998.
- Gurfinkel E, Bozovich G, Beck E, Testa E, Livellara B, Mautner B. The impact of inflammation and infection on the clinical outcome of patients with symptomatic coronary atherosclerosis disease. The final report of the ROXIS study. Eur Heart J 1999;20:121-127.
- Saikku P, Mattila K, Huttunen MS, Leinonen MS, Nieminen MS, Ekman MR et al. Serological evidence of an association of a novel Chlamydia, TWAR, with chronic coronary heart disease and acute myocardial infarction. Lancet 1988;2:983-986.
- 46. Ramírez JA. Isolation of Chlamydia pneumoniae from the coronary atherosclerosis. The Chlamydia pneumoniae/ Atherosclerosis Study Group. Ann Intern Med 1996;125:979-982.
- Danesh J, Collins R, Peto R. Chronic infections and coronary heart disease: is there a link? Lancet 1997;350:430-436.
- Kuo CC, Grayston JT, Campbell LA, Goo YA, Wissler RW, Benditt EP. Chlamydia pneumoniae (TWAR) in coronary arteries of young adults (15-34 years old). Proc Natl Acad Sci USA 1995;92:6911-6914.
- Libby P, Egan D, Skarlatos S. Roles of infectious agents in atherosclerosis and restenosis- An assessment of the evidence and need for future research. Circulation 1997;96:4095-4103.
* Tomado de: Rev Esp Cardiol 2001;54(3):383-92

