










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Medicina
versión On-line ISSN 1561-302X
Rev cubana med v.45 n.3 Ciudad de la Habana jul.-sep. 2006
Hospital Clinicoquirúrgico "Hermanos Ameijeiras
Servicio de Cardiología
Fisiopatología de los síndromes coronarios agudos
Dr. Javier Almeida Gómez1 y Dr. Orlando Álvarez Toledo2
Resumen
En la angina estable crónica, los episodios isquémicos (anginosos o silentes) ocurren fundamentalmente por un incremento de las demandas miocárdicas de oxígeno que exceden la capacidad de aumentar el flujo coronario por una estenosis significativa (usualmente mayor del 70 %) de una arteria epicárdica, o sea, la relación oferta y demanda de O2, se rompe al aumentar las demandas en relación con una oferta de flujo coronario reducida y relativamente fija. Por el contrario, en los síndromes coronarios agudos: angina inestable, infarto agudo del miocardio y muerte súbita de causa isquémica, la complicación primaria es, usualmente, una disminución súbita del flujo coronario secundario a la ruptura de una placa arteriosclerótica con la consecuente formación de un trombo. Teniendo en cuenta que el síndrome coronario agudo constituye la forma más severa de la enfermedad coronaria, que es un campo de investigación muy activo que tiene implicaciones terapéuticas y una patogenia cambiante, se realizó este artículo para comprender mejor los mecanismos involucrados en el síndrome coronario agudo y lograr mejor control y tratamiento.
Palabras clave: Síndrome coronario agudo, fisiopatología.
Existen varias diferencias patogénicas que determinan el tipo específico de síndrome coronario agudo (SCA) que se desarrolla.
La severidad del daño de la placa, la carga trombótica en el momento de la ruptura y asociado a ello el vasospasmo, desempeñan un papel fisiológico determinante en la presentación clínica de los diferentes síndromes coronarios agudos.1,2
Esencialmente, ello va a depender de varios factores, entre los que se encuentran:
- Severidad del daño de la placa: la cual puede ser desde una ligera fisura hasta una ruptura profunda con formación de una úlcera extensa.
- Factores trombogénicos locales y sistémicos en el momento de la ruptura de la placa: estos determinan la magnitud del trombo que se forme y su persistencia en el lugar de la placa inestable.
Factores trombogénicos locales
- Tamaño de la rotura.
- Sustrato trombogénico.
- Trombo residual.
- Irregularidades de la superficie.
- Grado de estenosis.
- Vasoconstricción.
Factores trombogénicos sistémicos
- Concentraciones elevadas de catecolaminas.
- Concentraciones de colesterol, lipoproteína (a) e hiperhomocisteinemia.
- Alteraciones en la fibrinólisis, función plaquetaria, coagulación, concentraciones de fibrinógeno y factor VII.
Vasoconstricción
Forma parte de los factores trombogénicos locales y contribuye a la isquemia, no solo por producirse en el sitio de la placa inestable o en zonas adyacentes a ellas, sino al nivel de la microcirculación coronaria. El vasospasmo en la zona de la ruptura de la placa puede producir oclusión coronaria intermitente, lo cual fue demostrado en estudios donde se utilizó estreptoquinasa intracoronaria. Las causas de esta vasoconstricción coronaria están relacionadas con un factor dependiente del endotelio (disfunción endotelial), así como otros factores dependientes de plaquetas y trombina.3-5
El endotelio normal libera sustancias con acción vasodilatadora y otras con efecto vasoconstrictor, entre los primeros se encuentra la prostaciclina, el factor relajante derivado del endotelio (óxido nítrico) y factor hiperpolarizante derivado del endotelio. El factor vasoconstrictor fundamental segregado por el endotelio es la endotelina 1. En condiciones fisiológicas hay un predominio de las sustancias vasodilatadores, especialmente dependientes de la liberación de ON. La disfunción endotelial comienza, entre otras causas, por infiltración de lípidos en el endotelio vascular (LDL-C). En los SCA, en la zona de la lesión culpable y adyacente se produce un aumento de la liberación de endotelina 1 y disminuye la liberación de sustancias que median vasodilatación dependiente del endotelio, el resultado de ello es un vasospasmo coronario en la zona de la lesión, además de un aumento del tono vasomotor al nivel de la microcirculación. Dos potentes vasoconstrictores coronarios, el tromboxano A2 y la serotonina, son liberados por las plaquetas adheridas al trombo. Del mismo modo, la trombina adherida al trombo es también un potente vasoconstrictor coronario que actúa directamente sobre las fibras musculares lisas.6
Por tanto, en los SCA se produce una obstrucción al libre flujo coronario por una factor fijo o anatómico dado por los cambios geométricos que sufre la placa al inestabilizarse así como el trombo sobreañadido, y por un factor dinámico que produce vasoconstricción tanto en el sitio de la lesión como distal a ella.3
Factores trombogénicos locales
Tamaño de la rotura
Experimentalmente, cuando se exponen las capas subendoteliales de la pared arterial a la sangre circulante en condiciones que simulen una estenosis coronaria significativa (fuerzas de cizalladura elevadas) se induce la agregación plaquetaria, pero el trombo resultante es lábil y se puede desprender fácilmente del sustrato y dejar pequeños trombos murales.6
Sin embargo, cuando se exponen capas arteriales más profundas se produce un trombo plaquetario denso y difícilmente desprendible del sustrato. Desde un punto de vista clínico, la erosión superficial de una placa producirá un estímulo trombogénico relativamente limitado y la respuesta trombótica puede no provocar más que un trombo mural que pueda posteriormente organizarse y contribuir al crecimiento asintomático de la lesión, pero un daño más profundo en la lesión provocará una respuesta trombótica mayor con compromiso del flujo distal y desencadenar un cuadro isquémico coronario.
Sustrato trombogénico
La respuesta trombótica también se ve influenciada por los diferentes componentes de la lesión aterosclerótica que queden expuestos tras la erosión o rotura de la placa. El núcleo lipídico es hasta 6 veces más trombogénico que la matriz rica en fibras de colágeno. Las placas que contengan un núcleo lipídico amplio no son sólo las más vulnerables a la rotura, sino que, además, una vez rotas son también las más trombogénicas. Aunque se desconoce el componente responsable de la alta trombogenicidad del núcleo lipídico, se sospecha que la existencia de factor hístico procedente de la degradación de macrófagos cargados de lípidos puede ser clave en el inicio de la coagulación tras la rotura. La presencia de factor hístico en el núcleo lipídico se ha demostrado con inmunohistoquímica en placas obtenidas tras endarterectomía carotídea y, lo que es más importante, utilizando un test funcional se ha demostrado que el factor hístico encontrado en el núcleo lipídico es activo y capaz de poner en marcha la cascada de la coagulación y la agregación plaquetaria.7-10
Grado de estenosis
La respuesta trombótica tras la rotura de una lesión aterosclerótica dependerá también del grado de estenosis y de los cambios geométricos bruscos que sigan a la rotura. La existencia de fuerzas de cizalladura elevadas en las estenosis predispone a la agregación plaquetaria y al depósito de fibrinógeno en la zona del daño vascular. Además, si la rotura sucede en el vértice de la estenosis, el trombo será más rico en plaquetas y menos susceptible de ser lisado por agentes trombolíticos que el trombo que se forme en la zona distal de la estenosis.
Irregularidades de la superficie
Además del grado de estenosis y la composición de la placa, la rotura de la cápsula fibrosa provoca irregularidades dentro de la luz vascular que pueden de por sí estimular el desarrollo de trombosis. En estudios de perfusión ex vivo de placas ateroscleróticas aórticas se ha demostrado que la presencia de flaps microscópicos, disecciones, fisuras y rugosidades en el lugar de la rotura también influyen en la trombogenicidad de la pared. A medida que estas irregularidades son mayores se producen alteraciones del flujo sanguíneo sobre la superficie con un flujo más turbulento que probablemente contribuyan a incrementar el depósito de plaquetas y fibrinógeno.7,10
Trombo residual
Después de la lisis de un trombo arterial existe una predisposición a la trombosis recurrente y reoclusión. El trombo residual puede sobresalir hacia la luz e incrementar la estenosis y facilitar así el depósito de plaquetas y fibrinógeno. El trombo, además, es muy trombogénico en sí mismo. La exposición de la trombina contenida en el trombo al torrente circulatorio estimula la agregación plaquetaria, activación de la coagulación y retrombosis. Experimentalmente, sólo la utilización de agentes directos antitrombina ha logrado abolir el crecimiento del trombo tras la rotura vascular.
Factores trombogénicos sistémicos
Además de los factores locales, existen evidencias tanto experimentales como clínicas de que determinados factores sistémicos puedan inducir estados de hipercoagulabilidad que potencien la trombosis tras la rotura de una lesión aterosclerótica.
Concentraciones elevadas de catecolaminas
La activación plaquetaria y la generación de trombina pueden verse potenciadas por el sistema nervioso simpático. El efecto trombogénico de la adrenalina parece estar relacionado con la activación de otros factores como la serotonina, el adenosindifosfato (ADP) o el tromboxano A2. Sin embargo, el efecto de la noradrenalina en la función plaquetaria es más controvertido. Un estudio experimental reciente ha demostrado efectos contrapuestos entre adrenalina y noradrenalina en la trombosis coronaria. Parece ser que actividades como fumar o el estrés mental, en las que predomina un aumento de la adrenalina, estarían asociadas con mayor riesgo de trombosis, mientras que en actividades como el ejercicio, donde se eleva sobre todo la noradrenalina, existiría una disminución del riesgo de trombosis.11-17
Concentraciones de colesterol, lipoproteína (a) y otras alteraciones metabólicas
La hipercolesterolemia se asocia con hipercoagulabilidad e incremento de la reactividad plaquetaria. El mecanismo por el que las concentraciones elevadas de colesterol influyen en la formación aguda de trombos se desconoce. Se sabe que la apo (a), que es una proteína presente en la lipoproteína (a), tiene una estructura muy similar a la del plasminógeno, y hay evidencias que sugieren que existe una inhibición competitiva entre lipoproteína (a) y plasminógeno, lo cual predispone a aquellos pacientes con concentraciones altas de lipoproteína (a) a presentar más complicaciones trombóticas. Además, un estudio angiográfico reciente ha demostrado que aquellos pacientes con progresión rápida de la enfermedad presentan más frecuentemente concentraciones elevadas de lipoproteína (a).6,14,15 Por otro lado, en los pacientes con angina inestable y en la fase aguda del infarto de miocardio se ha observado una disminución en las concentraciones de la apo A-1, proteína asociada a la superficie de las lipoproteínas de alta densidad (HDL [high density lipoprotein]), por lo que se ha sugerido que las HDL, además de intervenir en eliminar acumulaciones de colesterol de las lesiones, también pueden prevenir la formación de trombosis intracoronaria. En concreto, las HDL podrían estabilizar la prostaciclina a través de una función recientemente descubierta en la apo A-1, denominada factor estabilizador de la PGI2. Otras alteraciones metabólicas, como la diabetes, pueden aumentar la reactividad plaquetaria y estimular la coagulación, quizás a través de un aumento plasmático del cofactor de Von Willebrand u otros factores plasmáticos, o mediante alteraciones en el contenido lipídico de las membranas secundario a alteraciones en las lipoproteínas.
Alteraciones en la fibrinólisis, función plaquetaria, coagulación, concentraciones de fibrinógeno y factor VII
Todas las consideraciones anteriores sobre los efectos de las catecolaminas, colesterol o diabetes que incrementan la agregación plaquetaria y la coagulación y, consecuentemente, la incidencia de síndromes isquémicos, indican que las alteraciones en la activación plaquetaria y la coagulación son en sí mismas factores de riesgo en pacientes con enfermedad cardiovascular.18-22
En efecto, se ha comprobado que un incremento en la agregación plaquetaria inducida por trombina puede ser un marcador de futuras complicaciones coronarias o progresión de la enfermedad. También se ha observado que hay pacientes en los que tras un período de estabilización, después de un proceso isquémico agudo, persiste una generación continua de trombina y se ha sugerido que esta actividad pudiera ser desencadenante de isquemias recurrentes. Otras proteínas hemostáticas como el fibrinógeno y el factor VII se han considerado como posibles factores trombogénicos. Varios estudios prospectivos en los que se ha demostrado que las concentraciones elevadas de fibrinógeno se asocian de forma independiente con enfermedad coronaria, en particular con la incidencia de infarto de miocardio. De igual forma, las concentraciones elevadas de factor VII se han asociado con una incidencia mayor de infarto de miocardio. Ambas proteínas se elevan progresivamente con la edad, la obesidad, la hiperlipemia, la diabetes, el tabaco o el estrés emocional, por lo que pueden ayudar a explicar parcialmente el efecto de otros factores de riesgo sobre la enfermedad aterosclerótica.23,24
Otros factores
En los síndromes coronarios agudos sin elevación del segmento ST, la elevación de la troponina y la complejidad angiográfica de la lesión coronaria causante se relacionan con el incremento de la PCR. Este hallazgo apoyaría que la actividad inflamatoria, tanto de la pared vascular como del foco de necrosis, podría estar implicada en la patogenia de la elevación de los marcadores de inflamación (fig. 1).

Fig.1. Las manifestaciones agudas de la cardiopatía isquémica parecen ser la consecuencia de una alteración inflamatoria. Una lesión aterosclerótica puede comenzar teniendo una superficie lisa y no complicada (A) y dar lugar a un cuadro clínicamente silente, para convertirse después en una lesión complicada, intensamente ulcerada y hemorrágica (B) acompañada por el desarrollo de un cuadro clínico agudo e inestable. El mecanismo de esta transformación está relacionado con la puesta en marcha de una respuesta inflamatoria asociada a la lesión vascular inicial.
En 1985, Bevilacqua y otros demostraron que las citocinas estimulaban el endotelio, que se hacía más adhesivo para los leucocitos. Esta observación desarrolló una revolución en el entendimiento de la patogenia de la inflamación, y estudios posteriores corroboraron que el endotelio es el centro de los acontecimientos que permiten el desarrollo de la lesión inflamatoria. La adhesión de los leucocitos circulantes al endotelio vascular es el paso fundamental para su extravasación durante la inflamación. Este proceso está mediado por la molécula de adhesión E-selectina. Las moléculas de adhesión endoteliales pueden desprenderse de la superficie celular a la circulación y sus valores solubles pueden reflejar su expresión sobre la superficie endotelial. La respuesta inflamatoria aguda es un importante componente en la patogenia del daño miocárdico durante el síndrome coronario agudo y la disfunción endotelial está especialmente relacionada con el reclutamiento de los leucocitos durante la formación de la lesión aterosclerótica (fig.2). En la práctica clínica, se ha demostrado la utilidad de la detección sérica de diferentes marcadores de la inflamación, como la proteína C reactiva, el amiloide A, la troponina T y las citocinas, como las interleucinas (IL) 1 y 6 asociadas a la patogenia del síndrome coronario agudo y su diagnóstico diferencial, por lo que algunos de ellos se han utilizado como marcadores diagnósticos y pronósticos.25-32
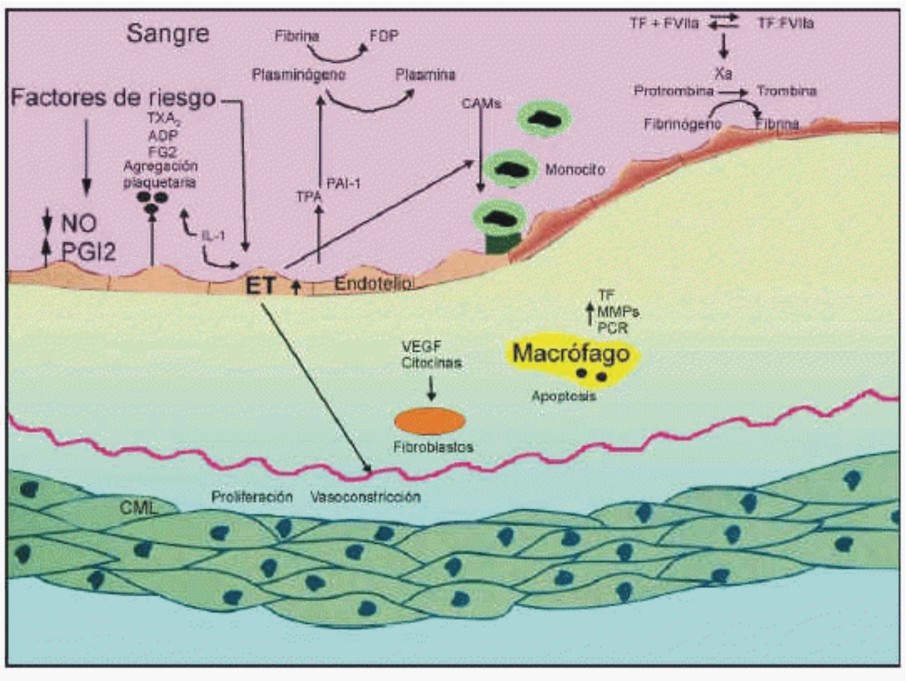
Fig.2. Disfunción endotelial avanzada. En la disfunción endotelial se produce vasoconstricción, las células endoteliales incrementan la expresión de VCAM e ICAM-1 que facilita la adhesión de monocitos y plaquetas a los vasos. Los mediadores inflamatorios causan apoptosis de las células endoteliales y las células musculares lisas, y producen proteinasas como las MMP-2 reguladas por las lipoproteínas oxidadas y las citocinas. Las MMP-2 contribuyen a activar la agregación plaquetaria, junto con los gránulos liberados por las propias plaquetas (TXA2, ADP y FG2). Las células apoptóticas liberan FT activado que desencadena la cascada de la coagulación. El FT interactúa con el FVII, activa el FX, que activa la conversión de protrombina a trombina. Finalmente, se genera fibrina que, junto con las plaquetas activadas, crean la estabilidad suficiente para la formación del trombo mural. El FT activado por los factores de riesgo sistémicos contribuye a la actividad procoagulante de las lesiones ateroscleróticas y a la formación del trombo.
Consecuencias clínicas de la rotura de la placa
No siempre la rotura de una lesión coronaria produce un cuadro de isquemia aguda; de hecho, se podría afirmar que esto es la excepción más que la regla. En estudios de autopsias se ha demostrado que existe rotura de lesiones coronarias hasta en el 9 % de los sujetos que han fallecido por causas no cardíacas y hasta en un 22 % de los sujetos con diabetes o hipertensión. Además, en la aorta abdominal se encuentran muy frecuentemente cápsulas fibrosas rotas con intensa actividad inflamatoria, y también se observan roturas asintomáticas de placas carotídeas hasta en un 20 % de las autopsias en pacientes ancianos.4 La mayoría de las roturas de placas provocarían la formación de un trombo mural, no oclusivo y asintomático, seguido de un proceso inflamatorio de reparación que pudiera contribuir al crecimiento rápido de la lesión (fase 3).13,18 Sin embargo, en estudios angiográficos recientes se ha observado de forma inequívoca que la progresión de las lesiones coronarias hacia formas avanzadas capaces de producir los síntomas suele ser impredecible y frecuentemente no lineal como cabría esperar de la acumulación de lípidos y de la proliferación celular. Frecuentemente, en los angiogramas aparecen nuevas lesiones severas en segmentos que estaban previamente sanos, y en las autopsias de pacientes fallecidos de cardiopatía isquémica suelen observarse lesiones con aspecto morfológico indicativo de la existencia de fisuras y trombosis en diferentes estadios de reparación y organización. En cualquier caso, aunque la mayoría de las roturas de lesiones ateroscleróticas pudieran ser pequeñas y contribuir a un crecimiento episódico de la lesión, ocasionalmente puede formarse un trombo capaz de comprometer el flujo arterial y provocar cuadros de isquemia aguda. En los cuadros de angina inestable, una pequeña erosión o fisura en una placa aterosclerótica puede producir cambios bruscos en la geometría de la estenosis y provocar cambios en el patrón de la angina. Episodios transitorios de oclusión trombótica podrían provocar angina de reposo. Este trombo suele ser lábil y la oclusión temporal, quizá no más de 15-20 min. Además, la vasoconstricción puede contribuir a reducir el flujo coronario distal de forma importante en los pacientes con angina inestable. En los casos de infarto de miocardio sin onda Q, un daño más severo de la lesión provocaría una oclusión trombótica más duradera, quizás hasta de 1 h. La cuarta parte de los pacientes con infarto sin onda Q puede tener la arteria responsable ocluida durante más tiempo, pero el territorio miocárdico suele estar irrigado por ramas colaterales.21,22
Además, la resolución de un posible vasospasmo asociado puede ser importante en la patogenia del infarto sin onda Q. En el infarto con onda Q, una rotura todavía mayor provoca la formación de un trombo de gran tamaño, estable y persistente que interrumpe de forma brusca el flujo coronario durante más de 1 h, produciendo la necrosis transmural del miocardio correspondiente. La lesión coronaria responsable del infarto es, con frecuencia, sólo de leve a moderadamente estenótica antes de que sobrevenga la oclusión. Es, pues, la rotura y trombosis más que la severidad de la lesión, el principal determinante de la oclusión aguda del vaso, y con mucha menor frecuencia la trombosis puede ser el resultado de una erosión superficial en una lesión severamente estenótica.
En resumen, la historia natural de los síndromes coronarios agudos es el reflejo de la evolución de la rotura y trombosis de la placa aterosclerótica. La estabilización correspondería a la reparación de la rotura, la acentuación de la angina correspondería a la presencia de trombos lábiles, el infarto de miocardio sin onda Q, a una oclusión más o menos transitoria, y el infarto transmural, a la existencia de un trombo oclusivo persistente.8
Es la composición de la placa más que el tamaño, el principal determinante de la vulnerabilidad, rotura y trombosis de las lesiones ateroscleróticas y, además, esta historia natural puede verse alterada por la presencia de circulación colateral o alteraciones en el tono vasomotor coronario.
Estructura de la placa arteriosclerótica coronaria
Cualquier explicación de los síntomas de la enfermedad arteriosclerótica debe comenzar en la placa (fig. 3).
Una placa arteriosclerótica coronaria está compuesta por un núcleo lipídico, separada del lumen vascular por una cápsula de tejido fibroso o fibromuscular, que a su vez está cubierto por endotelio vascular usualmente disfuncionante.
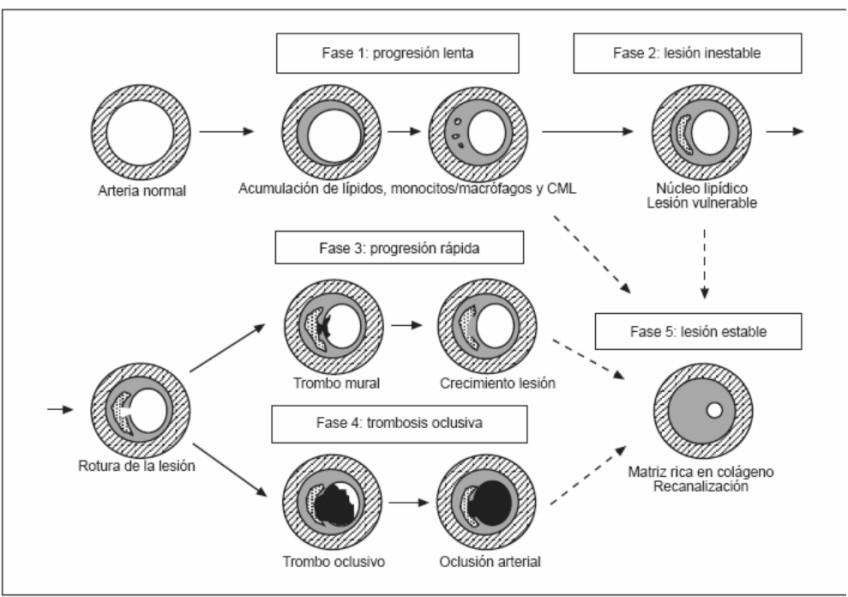
Fig. 3. Etapas o fases en el desarrollo de las lesiones ateroscleróticas definidas por el Comité de Lesiones Vasculares de la Sociedad Americana de Cardiología.
La cápsula de la placa tiene una elevada concentración de colágeno fibrilar tipo I densamente organizado. Dentro de las lagunas que forma el tejido conectivo hay células musculares lisas responsables de la producción de la matriz proteica del tejido conectivo incluyendo la colágena. El tejido de la cápsula presenta determinadas características morfológicas que sugieren que es capaz de soportar considerable estrés tensil sin fracturarse. El grosor de la cápsula en una misma placa varía ampliamente, pudiendo ser uniforme (delgada o gruesa), o por el contrario tener áreas gruesas y delgadas.21,22
El núcleo lipídico puede, en un extremo ocupar una proporción relativamente pequeña del contenido de la placa, o en el otro extremo ocupar un volumen considerable de la misma. El núcleo es esencialmente acelular y no contiene colágeno lo que implica que si los lípidos son removidos de la placa queda un espacio potencial en el interior. Existen además, placas que no tienen prácticamente contenido lipídico, en ellas predomina el tejido fibroso, lo cual ocurre sobre todo en placa que produce estenosis coronarias significativas. Estudios de trombogenicidad relativa de los diferentes componentes de la placa confirman que el núcleo lipídico es el sitio más activo que estimula la formación de un trombo.
Rodeando al núcleo lipídico hay numerosos macrófagos, su número varía ampliamente de una placa a otra aun en un mismo individuo. La función de los macrófagos será expuesta más adelante.
CML: Células musculares lisas
Formas de producirse la inestabilidad de la placa
Erosión o denudación del endotelio que cubre la cápsula fibrosa de la placa con la consecuente formación de un trombo.
Ruptura de la cápsula fibrosa de la placa, fundamentalmente de las placas blandas, con núcleos ricos en lípidos. En este caso la sangre penetra al núcleo de la placa formando un trombo (hemorragia intraplaca).
La relación entre ruptura de la placa y erosión endotelial aislada es de 3:1.
Mecanismo 1: Erosión o denudación del endotelio.
- Ocurre sobre todo en placas con estenosis severas.
- Más frecuente en la arteria descendente posterior.
- Se ve más en jóvenes y mujeres.
Al producirse la erosión endotelial se produce exposición de fibras colágenas y de factor hístico, lo que produce adhesión y activación plaquetaria. La propia pérdida del endotelio constituye un estímulo para la regeneración endotelial, el cual suele ser disfuncionante, con predisposición a la vasoconstricción. Si la denudación endotelial es amplia, además de la activación plaquetaria se produce depósito de fibrina con la formación de un trombo mayor.11,12
Mecanismo 2: Ruptura de la cápsula fibrosa de la placa con daño profundo de la íntima.
Al producirse la ruptura de la cápsula fibromuscular de la placa (que inevitablemente produce destrucción endotelial), la sangre penetra en el núcleo lipídico de la placa y se pone en contacto con las fibras colágenas y el factor hístico.
El grado de daño en la placa puede ser variable; en el mejor de los casos, la ruptura de la cápsula es pequeña y a pesar de que la placa se expande por la entrada de sangre al núcleo lipídico, el cambio en su geometría es discreto. En el otro extremo se encuentra la ruptura amplia de la cápsula fibrosa, que permite la entrada suficiente de sangre al núcleo lipídico para la formación de un trombo en expansión que se proyecta hacia la luz del vaso, produciendo rápidamente una estenosis significativa y limitando de forma importante el flujo distal.
Por otro lado, la ruptura de la cápsula puede ser múltiple con extrusión del contenido de la placa hacia el lumen vascular.
Estadios del proceso de ruptura de la placa
Un estadio inicial que se caracteriza por la entrada de sangre hacia el núcleo de la placa a través de la ruptura de la cápsula fibromuscular, a este estadio se le ha llamado hemorragia intraplaca, hematoma de la placa, disección hemorrágica o trombosis intraplaca. El componente fundamental del trombo intraplaca son las plaquetas, en mucha menor proporción, fibrina y eritrocitos.11,12
El siguiente estadio es la formación de un trombo en el área donde se rompe la cápsula, o sea, entre el núcleo de la placa y el lumen vascular. Este trombo, al cual se le denomina trombo mural o transicional, impide el ingreso de más sangre al núcleo de la placa (excepto cuando la ruptura de la cápsula es completa y se forma una úlcera que comunica directamente el núcleo lipídico con la luz vascular). Este trombo mural o transicional está en contacto directo con el flujo sanguíneo de la arteria por lo cual se pueden producir embolismos dístales predominantemente plaquetarios. El componente fundamental del trombo mural es la fibrina, pero la superficie externa está cubierta por una placa activa de plaquetas.
El estadio final lo constituye la formación de un trombo oclusivo que en su parte proximal está compuesto por fibrinas y plaquetas en iguales proporciones, pero inmediatamente distal a ella hay predominio de células rojas entremezcladas con una malla de fibrina.
En la angina inestable se produce ruptura de placa con formación de un trombo mural directamente expuesto al lumen vascular. La evolución ulterior del trombo mural se relaciona con el balance entre factores que promueven e inhiben la trombosis. La superficie activa del trombo mural está cubierta por una capa activa de plaquetas, debajo de la cual hay una densa capa de fibrina. La relativa ineficacia de los medicamentos trombolíticos sugiere que esta capa de fibrina es difícilmente asequible a estos medicamentos y que los fármacos antiplaquetarios desempeñan un papel fundamental al actuar sobre la superficie activa del trombo, evitando la progresión hacia un trombo oclusivo. Si en el momento de la fractura de la placa y formación del trombo mural predominan los factores que promueven la trombosis (factores trombogénicos locales y sistémicos), la tendencia será hacia la formación de un trombo oclusivo.
En la angina inestable (específicamente la angina de reposo), se pueden producir oclusiones totales transitorias entre 10 y 20 min. En el IMA transmural (IMA Q), la oclusión total es más persistente, por lo general mayor de 1 h y expresa el balance desfavorable entre factores que promueven e inhiben la trombosis a favor de los primeros, en este caso también tiene un peso importante la severidad del daño en la placa, por lo general mayor en el IMA Q, llega incluso a la ulceración de la placa, menor en la angina inestable e intermedio en el IMA no Q.15,16
Estudios de autopsias y de material obtenidos de aterotomias en pacientes con angina estable e inestable, muestran determinadas características de la placa que la hacen más susceptibles a la inestabilidad, entre ellas tenemos:
- Núcleo de la placa predominantemente lipídico, que ocupa gran porción del volumen total de la placa.
- Que la cápsula fibromuscular de la placa sea delgada.
- Incremento del número de macrófagos intraplaca (foam cell).
- Número reducido de células musculares lisas.
Si todos o la mayoría de estos factores coinciden en una misma placa, esta tiene un riesgo alto de inestabilizarse, sin embargo, cada uno de estos parámetros que determinan la vulnerabilidad de la placa son relativamente independientes, o sea, no están directamente vinculados, de este modo es posible que una placa tenga alto contenido lipídico, pero tenga una cápsula fibrosa gruesa y el contenido de macrófagos sea reducido, por lo que en general tendrá poco peligro de ruptura. Un paciente individual puede tener una o varias placas con características vulnerables para la ruptura y ocasionalmente puede presentar enfermedad multivaso con varias estenosis significativas y no presentar ninguna placa vulnerable.28-30
No existe en la actualidad ningún método clínico preciso para determinar el número ni las características de la placa aunque se están haciendo progresos en este sentido con introducción del US intracoronario.
Factores que promueven la extensión del trombo mural
- Espasmo local o distal.
- Aumento del fibrinógeno y/o factor VII.
- Aumento de la actividad del inhibidor tipo 1 del activador hístico del plasminógeno.
- Flujo reducido.
- Estenosis severa.
- Severidad del daño en la placa.
Factores que inhiben la extensión del trombo mural
- Flujo sanguíneo alto.
- Grado de lisis natural (prostaciclina, ON, etc.).
Mecanismos de ruptura de la placa
La fractura de cualquier tejido biológico depende de 2 variables: por un lado el estrés que actúa sobre un tejido y por otro, la resistencia mecánica de este tejido. Modelos computarizados que han estudiado el estrés circunferencial que actúa sobre la pared arterial en sístole, muestran que la distribución del mismo es desigual en presencia de un núcleo lipídico, este al ser blando y deformable no puede soportar el estrés circunferencial al que es sometido, entonces es redistribuido sobre la cápsula, especialmente sobre áreas relativamente pequeñas de la misma cuya posición va a estar en dependencia de la rigidez de la cápsula en relación con la íntima subyacente y por el ángulo circunferencial que ocupa el núcleo. De este modo, en determinados sitios de la cápsula el estrés circunferencial que actúa sobre ella llega a ser de 7 u 8 veces el normal, especialmente en aquellos lugares donde la cápsula es fina (se ha comprobado que en una misma placa el grosor de la cápsula es desigual y variable), el núcleo subyacente es rico en lípidos (cristales de colesterol) y la estenosis no es significativa. Estos sitios de la placa son mecánicamente ineficientes y por tanto más propensos a la ruptura.15-17
En cuanto a la fuerza o resistencia mecánica de la placa, se ha visto que la infiltración de macrófagos en la cápsula, así como la pérdida de colágeno en la misma, son 2 factores que contribuyen al debilitamiento de ella y por tanto, resulta más susceptible a la inestabilidad. La cápsula fibromuscular que cubre la placa es una estructura biológica en estado dinámico, en la cual existe un balance en la síntesis de fibras colágenas por las células musculares lisas y su degradación por metaloproteinasas liberadas por macrófagos (colagenasas, elastasas, gelatinasa B, etc.).25,26
De esta forma, la pérdida de células musculares lisas o la supresión de la síntesis de fibras colágenas por efecto de citoquinas (como interferón y liberadas por linfocitos) puede reducir la concentración de fibras colágenas en la cápsula y debilitarla.29 Cuando aumenta el número de macrófagos se produce un incremento localizado de su actividad proteolítica por la liberación de metaloproteinasas antes citadas, que digieren la matriz fibrosa de tejido conectivo que constituye la cápsula; estas áreas coinciden con los puntos de la cápsula donde es más probable la ruptura de la placa. Por lo tanto, hay un factor inflamatorio interno dentro de la propia placa (en el cual desempeñan un papel relevante los macrófagos activados) que inician el debilitamiento y la autodestrucción de la cápsula y que unido al factor externo (estrés circunferencial que actúa sobre la pared vascular), contribuyen a la inestabilidad de la placa.27,28,30,31
Dinámica de la formación del núcleo lipídico
El núcleo lipídico de la placa constituye el factor aislado más importante que influye en la vulnerabilidad de una placa para la ruptura. Está constituida por una acumulación de cristales de colesterol que forma aun espacio dentro de la matriz de tejido conectivo; los lípidos se derivan en su mayor parte de macrófagos muertos (foam cells) en las márgenes del núcleo lipídico acelular que liberan LDL-C oxidada que originalmente habían captado vía scavenger receptor. En una menor proporción, la LDL-C se encuentra ligada a proteoglicano y fibrinógeno sin haberse incorporado anteriormente a macrófagos.21-24
No está claro cómo los lípidos crean un defecto en la matriz de tejido conectivo. Pudiera tratarse simplemente de un hecho mecánico pasivo, en el cual los lípidos se abren paso separando las fibras colágenas. Sin embargo, estudios morfológicos sugieren qué mecanismo es más activo produciendo destrucción del tejido conectivo por metaloproteinasas liberados por macrófagos. Un hecho interesante es que los macrófagos raramente ocupan toda la circunferencia del núcleo lipídico y pueden estar totalmente ausentes en algunas placas con importante contenido lipídico.23,24
Summary
Physiopathology of the acute coronary syndromes
In the stable chronic angina, the ischemic episodes (anginous or silent) result mainly from a rise of the myocardial demand of oxygen exceeding the capacity of increasing the coronary flow due to a significant stenosis (usually over 70 %) of an epicardial artery, that is, the relation between the offer and the demand of O2 is broken on increasing the myocardial demands in relation to a reduced and relatively fixed offer of coronary flow. On the contrary, in the acute coronary syndromes (unstable angina, acute myocardial infarction and sudden death of ischemic cause) the primary complication is usually an abrupt decrease of the coronary flow secondary to the rupture of an atherosclerotic plaque with the subsequent formation of a thrombus. Taking into account that the acute coronary syndrome is the most severe form of the coronary disease,and that it is a very active research field with therapeutic implications and a changing pathogeny, this article was made to understand better the mechanisms involved in the acute coronary syndrome and to attain a more effective control and treatment.
Key words: Acute coronary syndrome, physiopathology.
Referencias bibliográficas
1. Fuster V, Badimon L, Badimon JJ, Chesebro JH. The pathogenesis of coronary artery disease and the acute coronary syndromes (part I). N Engl J Med.1992; 326:242-50, 310-8.
2. Fisiopatología de la angina inestable. Papel de la rotura y trombosis de la placa aterosclerótica. Rev Esp Cardio 1999; 52 [Supl 1]:3-12.
3. Tabata H, Mizuno K, Arakawa K, Satomura K, Shibuya T, Kurita A, et al. Angioscopic identification of coronary thrombus in patients with postinfarction angina. J Am Coll Cardiol. 1995;25:1.282-1.285.
4. Shilpesh SP, Thiagarajan R, Willerson JT, Yeh E. Inhibition of a4 integrin and ICAM-1 markedly attenuate macrophage homing to atherosclerotic plaques. Circulation. 1998;97:75-81.
5. Endemann G, Stanton LW, Madden KS, Bryant CM, White RT, Protter AA. CD36 is a receptor for oxidized low density lipoprotein. J Biol Chem. 1993;268:1.811-1.816.
6. Nozaki S, Kashiwagi H, Yamashita S, Nakagawa T, Kostner B, Tomiyama Y, et al. Reduced uptake of oxidized low density lipoproteins in monocyte-derived macrophages from CD36-deficient subjets. J Clin Invest. 1995;96:1.856-1.865.
7. Ramprasad MP, Terpstra V, Kondratenko N, Quehenberger O, Steinberg D. Cell surface expresion of mouse microsialin and human CD68 and their role as macrophage receptors for oxidized low density lipoprotein. Proc Natl Acad Sci USA. 1996;93:14.833-14.838.
8. Lee RT, Berditchevski F, Cheng GC, Hemler ME. Integrin-mediated collagen matrix reorganization by cultured human vascular smooth muscle cells. Circ Res. 1995;76:209-14.
9. El manejo del síndrome coronario agudo sin elevación del segmento ST. Aceptar la diversidad puede ser importante. Rev Esp Cardiol 2005;58:235-7. ISSN: 1579-2242.
10. Llorente V, Badimon L. Bases celulares y moleculares de la acumulación de colesterol en la pared vascular y su contribución a la progresión de la lesión aterosclerótica. Rev Esp Cardiol. 1998;51:633-41.
11. Loree HM, Tobias BJ, Gibson LJ, Kamm RD, Small DM, Lee RT. Mechanical properties of model atherosclerotic lesion lipid pools. Arterioscler Thromb. 1994;14:230-4.
12. Richardson RD, Davies MJ, Born GVR. Influence of plaque configuration and stress distribution on fissuring of coronary atherosclerotic plaques. Lancet. 1989;2:941-4.
13. Valor pronóstico del fibrinógeno en pacientes ingresados con sospecha de angina inestable o infarto de miocardio sin onda Q. Rev Esp Cardiol. 2002;55:622-30. ISSN: 1579-2242.
14. Shah PK, Falk E, Badimon JJ, Fernández-Ortiz A, Mailhac A, Villareal-Levy G, et al. Human monocyte-derived macrophages induce collagen breakdown in fibrous caps of atherosclerotic plaques: potential role of matrix-degrading metalloproteinases and implications for plaque rupture. Circulation. 1995;92:1.565-1.569.
15. Moreno P, Falk E, Palacios IF, Newell JB, Fuster V, Fallon JT. Macrophage infiltration in acute coronary syndromes: implications for plaque rupture. Circulation. 1994;90:775-8.
16. Geng Y, Wu Q, Muszynski M, Hansson GK, Libby P. Apoptosis of vascular smooth muscle cells induced by in vivo stimulation with interferon-g, tumor necrosis factor-a, and interleukin-1b. Arterioscler Thromb Vasc Biol. 1996;16:19-27.
17. Bennett MR, Evan GI, Schwartz SM. Apoptosis of rat vascular smooth muscle cells is regulated by p53-dependent and -independent pathways. Circ Res. 1995;77:266-73.
18. Ritmo luz/oscuridad de las citocinas proinflamatorias en el infarto agudo de miocardio. Rev Esp Cardiol. 2003; 56:555-60. ISSN:1579-2242.
19. Ip JH, Fuster V, Badimon L, Badimon J, Taubman MB, Chesebro JH. Syndromes of accelerated atherosclerosis: role of vascular injury and smooth muscle cell proliferation. J Am Coll Cardiol. 1990;15:1.667-1.687.
20. Presente y futuro del tratamiento antitrombótico en el síndrome coronario agudo. Rev Esp Cardiol. 2003;56:115-120. ISSN: 1579-2242.
21. Indicaciones de revascularización: aspectos clínicos. Rev Esp Cardiol. 2005;58:198-216. ISSN: 1579-2242.
22. Papel de los factores de riesgo en la trombogenicidad sanguínea y los síndromes coronarios agudos. Rev Esp Cardiol. 2003;56:1001-9. ISSN: 1579-2242.
23. Balbay Y, Tikiz H, Baptiste RJ, Ayaz S, Sasmaz H, Korkmaz S. Circulating interleukin-1 beta, interleukin-6, tumor necrosis factor-alpha, and soluble ICAM-1 in patients with chronic stable angina and myocardial infarction. Angiology. 2001;52:109-14.
24. Relación de los valores de proteína C reactiva con los hallazgos angiográficos y los marcadores de necrosis en el síndrome coronario agudo sin elevación del segmento ST. Rev Esp Cardiol. 2004;57:382-387. ISSN: 1579-2242.
25. Papel de los antiinflamatorios en el tratamiento de los síndromes coronarios agudos. De la ateroinflamación a la aterotrombosis. Rev Esp Cardiol. 2003;56:9-15. ISSN:1579-2242.
26. Moléculas de adhesión endoteliales ICAM-1, VCAM-1 y E-selectina en pacientes con síndrome coronario agudo. Rev Esp Cardiol. 2003;56:137-44. ISSN: 1579-2242.
27. Sanchis J, Bodí V, Llácer A, Fácila L, Núñez J, Bertomeu V, et al.. Uselfulness of concomitant myoglobin and troponin elevation as biochemical marker of mortality in non-ST-segment elevation acute coronary syndromes. Am J Cardiol. 2003;91:13-6.
28. Marcadores biológicos de necrosis miocárdica. Rev Esp Cardiol. 2003;56:703-720. ISSN: 1579-2242.
29. Marcadores de inflamación y estratificación de riesgo en pacientes con síndrome coronario agudo: diseño del estudio SIESTA (Systemic Inflammation Evaluation in patients with non-ST segment elevation Acute coronary syndromes). Rev Esp Cardiol. 2003;56:389-395. ISSN: 1579-2242.
30. Relación de los valores de proteína C reactiva con los hallazgos angiográficos y los marcadores de necrosis en el síndrome coronario agudo sin elevación del segmento ST. Rev Esp Cardiol. 2004;57:382-7.
31. Inflamación y síndrome coronario agudo. Rev Esp Cardiol. 2001;54:1135-1140. ISSN: 1579-2242.
32. Schieffer B, Schieffer E, Hilfiker-Kleiner D, Hilfiker A, Kovanen PT, Kaartinen M, et al. Expression of angiotensin II and interleukin 6. En: Pérez-Fernández R, Kaski JC. Interleucina-10 y enfermedad coronaria. Rev Esp Cardiol. 2002;55:738-50.
Recibido: 9 de febrero de 2006. Aprobado: 6 de abril de 2006.
Dr. Javier Almeida Gómez. Hospital Clinicoquirúrgico Hermanos Ameijeiras. San Lázaro No. 701 entre Belascoaín y Marqués González, Centro Habana, Ciudad de La Habana, Cuba.
1Especialista de I Grado en Medicina General Integral. Residente de 3er. Año en Cardiología.
2Especialista de I Grado en Cardiología. Jefe de la Unidad de Cuidados Coronarios.

