










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Medicina
versión On-line ISSN 1561-302X
Rev cubana med v.48 n.3 Ciudad de la Habana jul.-sep. 2009
PRESENTACIÓN DE CASOS
Enfermedad de Rendu-Osler-Weber
Rendu-Osler-Weber syndrome
Gloria Astencio RodríguezI; Onix César Garib AlpízarII; José Napoleón Ventura BlancoIII; Boris L. Torres CuevaIV; (†) Nancy de León Rubio; Jorge Luis HernándezV
IEspecialista de II Grado en Gastroenterología. Master en Bioética. Profesora Auxiliar. Hospital Clinicoquirúrgico "Hermanos Ameijeiras". La Habana, Cuba.
IIEspecialista en Medicina General Integral. Residente de 3er Año en Gastroenterología. Hospital Clinicoquirúrgico "Hermanos Ameijeiras". La Habana, Cuba.
IIIResidente de 3er Año en Gastroenterología. Hospital Clinicoquirúrgico "Hermanos Ameijeiras". La Habana, Cuba.
IVEspecialista de I Grado en Imagenología. Profesora Auxiliar. Hospital Clinicoquirúrgico "Hermanos Ameijeiras". La Habana, Cuba.
VLicienciado en Radiología. Hospital Clinicoquirúrgico "Hermanos Ameijeiras". La Habana, Cuba.
(†) Especialista de II Grado en Imagenología. Profesora Auxiliar. Hospital Clinicoquirúrgico "Hermanos Ameijeiras". La Habana, Cuba.
RESUMEN
La telangiectasia hemorrágica hereditaria o enfermedad de Rendu-Osler-Weber es autosómica dominante, se caracteriza por la presencia de múltiples telangiectasias en piel y mucosas asociadas a malformaciones arteriovenosas de distintos órganos. Con un diagnóstico y tratamiento precoces es posible mejorar el pronóstico, tanto la calidad como la expectativa de vida del paciente. El tratamiento del enfermo debe ser individualizado y realizar despistaje para malformaciones vasculares tanto a él como a sus familiares de primer grado, ya que puede cursar sin síntomas. Se presentó el caso de un adulto del sexo masculino hospitalizado en el Servicio de Gastroenterología del Hospital Clinicoquirúrgico "Hermanos Ameijeiras" por episodios recurrentes de sangrado digestivo alto, se le realizaron estudios vasculares que demostraron las lesiones típicas de la enfermedad, se trató mediante embolización selectiva, con la que se logró buena evolución posterior. Palabras clave: Telangiectasia hemorrágica hereditaria, Rendu-Osler-Weber, malformaciones arteriovenosas.
ABSTRACT
The hereditary hemorrhagic telangiectasia or Rendu-Osler-Weber is autosomal dominant, characterized by presence of multiple telangiectasias present in skin and mucosa associated with arteriovenous malformations of different organs. Prognosis although with an early diagnosis and treatment could be improve the patient's quality and expectation of life. Treatment must to be individualized and to carry out screening for vascular malformations in patient and its first degree relatives since it may be present without symptoms. This is the case of male patient admitted in Gastroenterology Service of "Hermanos Ameijeiras" Clinical Surgical Hospital due to recurrent episodes of high digestive bleeding. Vascular studies were carried out showing the typical lesions of disease. Treatment included a selective embolization achieving a subsequent good course.
Key words: Hereditary hemorrhagic telangiectasia, Rendu-Osler-Weber, arteriovenous malformations.
INTRODUCCIÓN
La telangiectasia hemorragica hereditaria (THH) o enfermedad de Rendu-Osler-Weber fue descrita por primera vez en el año 1876, por John Wickham Legg y, posteriormente, por Henri Jules Rendu, en 1896. En el año 1901, William Osler describió 3 pacientes que padecían una forma familiar rara de epistaxis (hemorragias nasales) recurrente, asociada con telangiectasias (dilatación de los vasos sanguíneos de tamaño muy pequeño que daba lugar a manchas de color púrpura con aspecto de araña) en piel y mucosas. Se trata de una angiopatía neoformativa de telangiectasias circunscritas que, al romperse, determinan síndromes hemorrágicos locales.
La THH es infrecuente, se estima una prevalencia de 2 casos por 100 000 personas, es mayor en algunas áreas geográficas, como en la isla danesa de Fyn, las Antillas Danesas y en algunas regiones de Francia. Esta enfermedad afecta principalmente pacientes caucásicos, aunque existen reportes ocasionales en pacientes asiáticos y árabes. Afecta por igual a ambos sexos, comienza más frecuentemente durante la pubertad o adultez, entre los 20 y los 40 años, aunque también puede presentarse en niños.1
La THH es una enfermedad vascular rara, congénita, que se caracteriza por la presencia de múltiples telangiectasias con tendencia a sufrir hemorragias localizadas principalmente en la nariz y en las vías urinarias, aunque también es posible que se desarrollen anomalías vasculares internas al nivel del cerebro, pulmones, garganta, laringe, tracto gastrointestinal, hígado, vejiga y vagina. Las telangiectasias suelen verse en labios, lengua y mucosa nasal, y también pueden afectar otras zonas como la cara y las orejas.1,2
Es hereditaria, con un rasgo autosómico dominante, por lo que basta que uno de los padres la padezca para que el hijo pueda resultar enfermo. Retrata de un gen individual, anormal en uno de los cromosomas autósomicos (1 de los primeros 22 cromosomas "no sexuales"). En su patogénesis están implicados 2 genes, THH1 y THH2, los cuales determinan 2 formas diferentes de una misma enfermedad. La variante THH1 se origina por mutaciones en el gen endoglina (ENG), localizado en el brazo largo del cromosoma 9 (9q33-q34.1), mientras que el THH2 es causado por mutaciones en el gen ALK1, localizado en el brazo largo del cromosoma 12 (12q11-q14).2,3
Se caracteriza por la presencia, desde el nacimiento, de múltiples telangiectasias en piel y mucosas (dilataciones venulares y capilares), propensión a hemorragias localizadas, principalmente nasales, urinarias (hematuria) y, con menor frecuencia, digestivas (gastrorragia) y respiratorias (hemoptisis).
PRESENTACIÓN DEL CASO
Paciente de 61 años, sexo masculino, raza mestiza con antecedentes de buena salud, que en 1978 presentó un episodio de melena en abundante cantidad, de la cual no se precisó la causa, acompañado en ocasiones de epistaxis. Aproximadamente, 20 d después presentó otro episodio similar al ya descrito, lo cual lo condujo a una anemia aguda (5,6 g/L) por lo que requirió transfusiones de glóbulos rojos. Al realizarle el examen físico se encontró, mucosas hipocoloreadas, en piel, una lesión de aspecto telangiectásico, papular, anular de 1 cm de diámetro en el pabellón auricular izquierdo, así como la presencia de un soplo sistólico en mesogastrio y flanco derecho del abdomen.
Exámenes complementarios realizados: Hb: 83 g/L, Hto: 0,25 /L, plaquetas: 433 x109/L, leucocitos: normales, VSG: normal, química sanguínea (BT, ácido úrico, colesterol, triglicéridos, amilasa, TGP, TGO, FAL, GGT, glucosa, PT, y albúmina): normales, coagulograma: normal, grupo sanguíneo: O positivo, VDRL: no reactivo.
Rayos X de tórax: no alteraciones pleuropulmonares ni cardiaórticas.
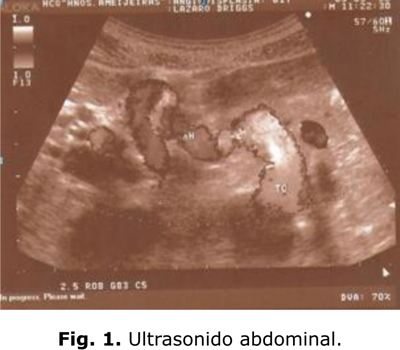
Ultrasonido abdominal: marcada dilatación del tronco celíaco con vasos de circulación colateral al nivel del hilio hepático (fig.1).
TAC de abdomen: se observa una alteración en la ecoestructura del hígado sobre todo al nivel de la vértebra L1, con dilatación de las venas intrahepáticas, hay calcificaciones anulares en proyección de la región pancreática, el hígado se define pobremente, se realiza examen con contraste endovenoso y se ve una gran dilatación del tronco celíaco y de la arteria hepática, que es muy tortuosa. El hepatograma no es homogéneo, se visualizan las venas intrahepáticas muy dilatadas. No hay alteraciones cráneo-encefálicas.
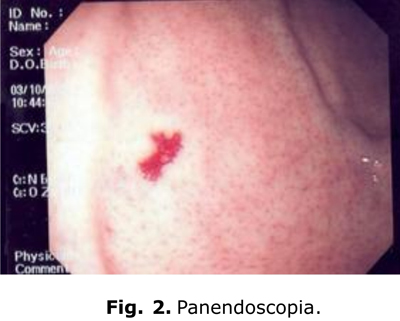
Panendoscopia: angiodisplasia gástrica, pangastritis eritematosa y bulbitis eritematosa (fig.2).
Ileocolonoscopia: normal, pólipo del sigmoide y polipectomía endoscópica satisfactoria.
TAC de pulmón-cráneo: se observa una lesión nodular de 1,87 cm. Con una densidad de 50 UH en hemitórax derecho retroesternal a la altura de la bifurcación de la tráquea. El nódulo es de contornos bien definidos. Hay una imagen nodular por delante del área cardíaca muy próxima a la línea media en íntima relación con vasos pulmonares que están dilatados y tortuosos. Al examen endovenoso se ve que la lesión en el estudio simple tiene una densidad de 31 UH y se eleva hasta 139 UH con el aspecto de una fístula arteriovenosa.
Se realiza arteriografía hepática, tronco celíaco, mesentérica y vasos del tubo digestivo (fig. 3).

Se logra comprobar la existencia de una lesión vascular de aspecto angiodisplásico que se nutre a partir de las primeras ramas mediales de la mesentérica superior y da la impresión que corresponde a ramas cólicas derechas con evidente extravasación del contraste, vasos tortuosos de drenaje venoso precoz con persistencia del propio material de contraste en ambos compartimientos intravascular y extravascular. La lesión mide aproximadamente 8,6 cm.
Al nivel del tronco celíaco, llama la atención su dilatación y de todo el eje hepático, con compromiso de la coronaria estomáquica, la cual está igualmente dilatada y con marcada tortuosidad de ambas arterias hepáticas derecha e izquierda, así como la hepática propia con un aspecto igualmente angiodisplásico, con una porta notablemente dilatada en vistas tardías. Existen 2 lesiones angiodisplásicas similares a la anteriormente descrita que se nutren fundamentalmente por la gastroduodenal y no se descarta la participación de la pancreático duodenal superior.
Tras el interrogatorio, el examen físico y los estudios realizados al paciente, el diagnóstico se concluyó como una enfermedad de Rendu-Osler-Weber, al cual se le practicó embolización selectiva de la rama aferente de la lesión angiodisplásica encontrada en el intestino, con lo cual se logró una evolución satisfactoria.
DISCUSIÓN
Los genes ENG y ALK1 codifican el factor de crecimiento transformante tipo b (TGFb) expresado en las células endoteliales. La angiogénesis es regulada por ENG y ALK1 como reguladores positivos durante las fases de activación y resolución. Durante las fases de activación, las células mesenquimales se diferencian a pericitos y células del músculo liso. Se forman nuevos vasos por la proliferación y migración de las células endoteliales inducidas por ALK1, mientras que ALK5 induce la fase de resolución del proceso. La angiogénesis requiere un balance positivo ALK1/ALK5 y el papel de la endoglina parece ser necesario para mantener el equilibrio. Por un mecanismo todavía desconocido, mutaciones genéticas en los genes ENG y ALK1 causan las alteraciones en la angiogénesis que determinan las telangiectasias y las malformaciones arteriovenosas. ALK1 es importante por su papel en la regulación de Efnb2, marcador molecular arterial. Todavía no se conoce con exactitud el mecanismo por el cual Efnb2 y ALK1 entran en relación, aunque es precisamente esta interacción la que determina las variaciones clínicas de THH1 y THH2. No obstante, la existencia de grupos familiares con síntomas compatibles con THH, pero sin las mutaciones genéticas, sugiere que otro gen todavía no identificado podría ser la causa de THH en esos casos. Estudios recientes en ratones muestran que el incremento del factor de crecimiento de endotelio vascular (VEGF) determina la formación de microvasos anormales en ENG heterocigotos. Sadick y otros encontraron que VEGF se expresa no sólo en al plasma, sino también en la mucosa nasal de los pacientes con THH.1-3
La base de los síntomas clínicos de la THH es la formación irregular de vasos sanguíneos. Las telangiectasias en la mucosa nasal y el sangrado nasal son los síntomas más tempranos y comunes de la THH como se refiere en este paciente. El 95 % de los pacientes afectados presentan epistaxis recurrente, que generalmente comienza a partir de los 12 años y se presenta con una frecuencia de 18 episodios por mes. El sangrado nasal severo puede causar anemia crónica, aunque a intervalos esporádicos, no requieren tratamiento. Generalmente, la frecuencia y la severidad del sangrado nasal se incrementan con la edad, aunque algunos pacientes no refieren estos cambios. Un porcentaje similar de pacientes presentan múltiples telangiectasias en manos, cara y cavidad oral, generalmente tras un período de epistaxis.3,4
Los pacientes afectados por THH pueden presentar telangiectasias gastrointestinales, de forma más frecuente en estómago y parte superior del duodeno; el 25 % de los afectados mayores de 60 años presenta sangrado gastrointestinal generalmente asociado a melena o anemia, en este paciente la manifestación clínica que motivó la hospitalización fue el sangrado digestivo recurrente, manifestado por episodios de melenas. El sangrado es lento y persistente, y puede empeorar con la edad. Las trombosis o embolias son complicaciones de las malformaciones arteriovenosas y pueden aumentar con el paso del tiempo.4,5
El compromiso hepático en pacientes con THH resulta principalmente de cortocircuitos entre la arteria hepática y las venas hepáticas. Las manifestaciones clínicas del compromiso hepático en estos pacientes van a depender del tipo y del tamaño del cortocircuito y de los efectos de la alteración de la irrigación hepática. La mayoría de los pacientes tienen una circulación hiperdinámica que resulta de los cortocircuitos arteriovenosos, portovenosos o de ambos, situación hemodinámica muy similar a la encontrada en nuestro caso. Un gran cortocircuito de izquierda a derecha puede resultar en una insuficiencia cardíaca de alto flujo. Los cortocircuitos secundarios a malformaciones entre la vena porta y la hepática pueden producir encefalopatía hepática luego de un sangrado en el tracto gastrointestinal.3,4
Malformaciones entre la arteria hepática y la vena porta pueden producir hipertensión portal con várices esofágicas. No se ha reportado sangrado de malformaciones arteriovenosas (MAV) hepáticas. Debe sospecharse la presencia de una MAV hepática en pacientes con hepatomegalia o con un soplo en el hígado, como fue encontrado en nuestro paciente. La TAC y la ecografía Doppler color son métodos sensibles y no invasivos para detectarlas. También puede utilizarse la angiografía en el estudio de estas lesiones. En los exámenes de laboratorio pueden pesquisarse aumentos discretos en los niveles de gamma glutamil transpeptidasa y fosfatasa alcalina, así como también una colestasia leve. La biopsia hepática es innecesaria, ya que no permite detectar la presencia de compromiso en este órgano.5-7
Las malformaciones arteriovenosas pulmonares ocurren aproximadamente en el 30 % de los individuos con THH. EL 30-40 % de pacientes con MAV pulmonares presentan alteraciones del sistema nervioso central, con complicaciones tromboembólicas como infarto, absceso cerebral o ataques isquémicos transitorios por la comunicación sanguínea. Las mujeres gestantes con MAV sin tratamiento presentan mayor riesgo de hemorragia pulmonar. En nuestro caso se encontró una MAV pulmonar sin manifestaciones clínicas hasta la fecha.
Las MAV del sistema nervioso central pueden ser congénitas En el 10 % de los pacientes con THH están presentas las MAV cerebrales, las cuales pueden aparecer a cualquier edad en forma de infartos, dolores de cabeza o hemorragia intracraneal. El 1 % presenta MAV espinales que pueden causar hemorragia subaracnoidea, mielopatía progresiva, dolor radicular o alteraciones en esfínteres. Es posible encontrar comunicaciones hepáticas con alta tasa de fracaso cardíaco, hipertensión portal, enfermedad biliar y encefalopatía portosistémica. Un estudio reciente ha identificado por tomografía computarizada anormalidades hepáticas en el 78 % de pacientes con THH, inclusive en casos asintomáticos.2,3,8
El diagnóstico inicial de THH continúa basándose en la presencia de signos clínicos compatibles junto con la historia familiar. Para el diagnóstico molecular es necesario secuenciar las regiones codificantes completas de los genes ALK1 y ENG, a pesar de que sólo es posible diagnosticar THH en el 70 % de los casos, ya que se han detectado nuevas variaciones en secuencias de desconocida significancia clínica. El test genético no es positivo en el 100 % de los pacientes con THH, es posible encontrar mutaciones diferentes en el mismo grupo familiar. Estudios futuros aclararán los motivos de esta discrepancia. En nuestro caso, el diagnóstico se basó fundamentalmente en la clínica (APP, APF y EF) y en estudios de imágenes y endoscópicos.8
Para poder establecer el diagnóstico, los pacientes deben cumplir al menos 3 de los 4 criterios siguientes:1 1. Epistaxis. 2. Telangiectasias múltiples en localizaciones típicas (dedos, labios, cavidad oral, nariz, lechos subungueales). 3. Lesiones viscerales, que incluyen:
a. Telangiectasias gastrointestinales (40 %).
b. MAV pulmonares (30 %).
c. MAV hepáticas (30 %).
d. MAV cerebrales (5 a 11 %).
e. MAV medulares.
4. Historia familiar, con un pariente de primer grado con THH.
Considerando los criterios diagnósticos antes señalados y teniendo en cuenta los elementos clínicos con los que se nos presentó el caso y lo obtenido en los estudios realizados se confirmó el diagnóstico en nuestro paciente de enfermedad de Rendu-Osler-Weber.
El principal procedimiento adoptado en estos casos es el tratamiento sintomático del sangrado oral y nasal, junto con la prevención de las posibles complicaciones, como hemorragia interna, pulmonar y cerebral por MAV.
Todos los procedimientos quirúrgicos para tratar las MAV pulmonares, cerebrales y hepáticas son muy peligrosos por el alto riesgo de hemorragia, defectos neurológicos o inclusive muerte que entrañan. Por estas razones, no existen protocolos quirúrgicos para tratar las lesiones de THH, valorando individualmente el tratamiento a seguir en función del riesgo. El sangrado gastrointestinal se trata con suplementos de hierro, etinilestradiol/noretindrona, danazol, ácido aminocaproico o, más recientemente, por aplicación endoscópica del láser.9,10
La anemia puede ser controlada de forma oral o parenteral con hierro o bien con transfusiones sanguíneas. El mejor tratamiento de las epitaxis leves es la aplicación diaria de lubricantes nasales y, en caso de sangrado nasal moderado, es aconsejable el empleo del láser, aunque para las formas severas pueden ser necesarios injertos de piel. Las manifestaciones en la piel generalmente no requieren tratamiento, pero en caso de sangrado o por motivos estéticos pueden ser eliminadas con láser. Los pacientes deberán evitar los medicamentos que interfieran con la coagulación.10
El tratamiento endoscópico con láser o con coagulación no ha tenido buenos resultados, principalmente porque las lesiones del intestino delgado no son alcanzadas con el endoscopio. En algunos pacientes se ha realizado embolización o ligadura de la arteria hepática. Aunque este procedimiento disminuye los síntomas de falla cardíaca o de secuestro esplénico, puede producir necrosis hepáticas, biliares o ambas, por lo que debería ser usado cuidadosamente y sólo en circunstancias especiales. Algunos pacientes pueden requerir trasplante hepático.10,11
REFERENCIAS BIBLIOGRÁFICAS
1. Telangiectasia hemorrágica hereditaria (enfermedad de Rendu-Osler-Weber): a propósito de un caso. Dermatol Pediatr Lat. 2004;2(2):130-8.
2. Síndrome de Rendu-Osler-Weber o telangiectasia hemorrágica hereditaria (HHT): Descripción de dos casos y revisión de la literatura. Avances en Odontoestomatología. 2005;21(6):
3. McDonald J, Bayrak-Toydemir P. Hereditary hemorragic telangiectasia. Haematologica. 2005;90:728-32.
4. Cirulli A, Liso A, D'Ovidio F, Mestice A, Pasculli G, Gallitelli M, et al. Vascular endothelial growth factor serum levels are elevated in patients with hereditary hemorrhagic telangiectasia. Acta Haematol. 2003;110:29-32.
5. Xu B, Wu YQ, Huey M, Arthur HM, Marchuk DA, Hashimoto T, et al. Vascular endothelial growth factor induces abnormal microvasculature in the endoglin heterozygous mouse brain. J Cereb Blood Flow Metab. 2004;24:237-44.
6. Memeo M, Stabile Ianora AA, Scardapane A, Buonamico P, Sabba C, Angelelli G. Hepatic involvement in hereditary hemorrhagic telangiectasia. Abdom Imaging. 2004;29:211-20.
7. Stabile Ianora AA, Memeo M, Sabba C, Cirulli A, Rotondo A, Angelelli G. Hereditary hemorrhagic telangiectasia: multidetector row helical T assessment. Radiology. 2004;23:250-9.
8. Saluja S, White RI. Hereditary hemorrhagic telangiectasia ischemia: poverty amidst plenty? Radiology. 2004;230:25-7.
9. Boillot O, Bianco F, Viale JP, Mion F, Mechet I, Gille D, et al. Liver transplantation resolves the hyperdynamic circulation in hereditary hemorrhagic telangiectasia with hepatic involvement Gastroenterology. 1999;116:187-92. 11. Fuchizaki U, Miyamori H, Kitagawa S, Kaneko S, Kobayashi K. Hereditary haemorrhagic telangiectasia Kobayashi K. Hereditary haemorrhagic telangiectasia (Rendu-Osler-Weber disease). Lancet. 2003;362:1490-94. Recibido: 24 de febrero de 2009.
10. Garcia-Tsao G, Korzenik J,Young L, Henderson KJ, Jain D, Byrd B, et al. Liver diseasae in patients with Hereditary Hemorrhagic Telangiectasia. N Engl J Med. 2000;343:931-6.
Aprobado: 2 de junio de 2009.
Dra. Gloria Astencio Rodríguez. Hospital Clinicoquirúrgico "Hermanos Ameijeiras", San Lázaro No. 701 entre Belascoaín y Marqués González, Centro Habana, Ciudad de La Habana, Cuba. CP 10300.

