










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Medicina
versión On-line ISSN 1561-302X
Rev cubana med v.50 n.1 Ciudad de la Habana ene.-mar. 2011
PRESENTACIÓN DE CASOS
Ganglioneuroblastoma sacro
Sacral ganglioneuroblastoma
Eileen de la Nuez Cobas,I Natacha Berland de León,II Anisia Otaño RodríguezIII
I Máster en Atención Integral a la Mujer. Especialista de I Grado en Imaginología. Instructora. Facultad "Dr. Salvador Allende". La Habana, Cuba.
II Especialista de I Grado en Imaginología. Profesora Auxiliar. Hospital Clinicoquirúrgico "Hermanos Ameijeiras". La Habana, Cuba.
III Especialista de I Grado en Imaginología. Facultad "Dr. Salvador Allende". La Habana, Cuba.
RESUMEN
Se presentó un caso raro de ganglioneuroblastoma sacro en un paciente de 32 años de edad, con evolución tórpida. Se presentan datos clínicos, estudios imaginológicos e histológicos. Se realiza revisión del tema.
Palabras clave: Neuroblastoma, ganglioneuroblastoma, TC contrastado.
ABSTRACT
This is a uncommon case of sacral ganglioneuroblastoma in a patient aged 32 with a torpid evolution. Clinical data, imaging and histological studies are showed. A review of this subject is carried out.
Key words: Neuroblastoma, ganglioneuroblastoma, contrasted-CT.
INTRODUCCIÓN
El tumor desmoplásico de células redondas (DSRCT) constituye una entidad particular con una diferenciación divergente y una extremada agresividad. Afecta a niños y jóvenes adultos, la localización más frecuente es la cavidad intraabdominal, forma grandes masas tumorales que ocupan la pelvis y el retroperitoneo. Una de las principales características de este tumor es su naturaleza plurifenotípica, con expresión epitelial, neural y biogénica.1,2
La familia de los tumores neuroblásticos (neuroblastomas) constituye el tipo tumoral sólido extracraneal más frecuente diagnosticado en la infancia. Estos tumores, derivados de la cresta neural y localizados en glándula suprarrenal o en territorios extra-adrenales, presentan unas características clínicas y biológicas curiosas, existen casos con remisión espontánea, junto a otros con progresión tumoral, escasa respuesta a la terapia y muerte del paciente.3
Factores clínicos como la edad del paciente (pronóstico favorable en niños menores de 1,5 años) y el estadio tumoral (favorable en estadios localizados de la enfermedad), son criterios fundamentales para la programación del tratamiento.
Caso clínico
Paciente de 32 años que refiere hace unas semanas que defeca, con dificultad, heces acintadas y presenta dolor lumbar. Acude al cuerpo de guardia por retención urinaria. Al examen físico se constata globo vesical. Se realiza ecografía abdominal (figs. 1 y 2).
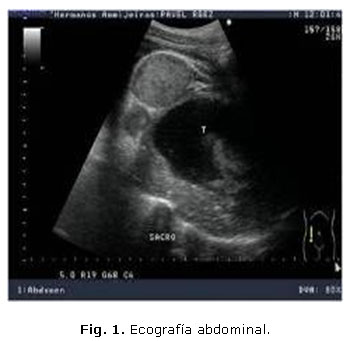
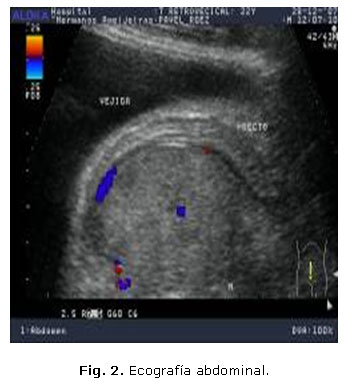
En ambas figuras se muestran imágenes de ultrasonido, modo B donde se observa una masa compleja, de 17,4 x 10,5 cm, con áreas de necrosis, por delante del sacro, que desplaza al recto y la vejiga hacia delante, el estudio con Doppler color muestra vascularización, sobre todo en su periferia.
Con los hallazgos anteriores se sugiere una TC con contraste vía oral y EV (figs. 3 y 4).
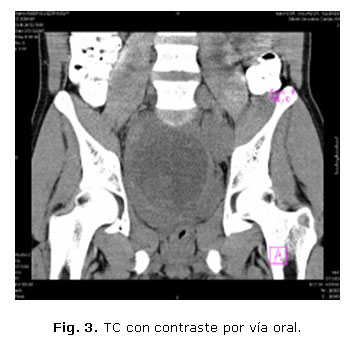
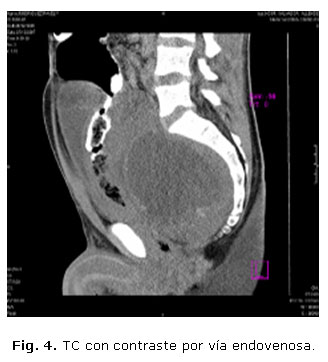
Las figuras 3 y 4 corresponden a imágenes de TC, vistas coronal y sagital, donde se observa una tumoración compleja en la excavación pélvica, con densidades bajas en su interior, asociada a áreas de necrosis y calcificaciones, situada por delante del sacro, que comprime la vejiga, colon izquierdo y recto hacia delante, estrechando la luz de este último.
Se realiza una CAAF guiada por ultrasonido, que informa posible ganglioneuroblastoma, se trata quirúrgicamente, se reseca el tumor y se comprueba con biopsia por parafina el diagnóstico (fig. 5).

DISCUSIÓN
Los neuroblastomas se desarrollan a partir del sistema nervioso simpático y aparecen con mayor frecuencia como masas abdominales, fundamentalmente en las glándulas suprarrenales como sitio primario, seguido de los ganglios simpáticos paraespinales, mediastino, pelvis, cabeza y cuello. Dichos tumores secretan altos niveles de dopamina y ácido homovalínico (HVA), lo cual trae como resultado bajos niveles de ácido vanilamandélico (VMA)/HVA y norepinefrina/dopamina.4,5 Los tumores con mejor evolución clínica secretan altos niveles de norepinefrina y VMA.
Los neuroblastomas hacen metástasis por vía linfática y hemática. Los sitios más comunes son: médula ósea (78 %), hueso (69 %), ganglios linfáticos (42 %), hígado (20 %), piel (2 %) y testículos (2 %). Las metástasis pulmonares y cerebrales son raras. En las masas tumorales, el componente glioneuromatoso predomina en más del 50 %.6 El pronóstico de los pacientes con ganglioneuroblastoma depende de varios factores, el más importante es la edad en la que se realizó el diagnóstico y el estadio. El pronóstico es más favorable en edades tempranas del diagnóstico y en bajos estadios de la enfermedad. La presencia del encogen N- myc en 25-30 % de los neuroblastomas se asocia con la progresión de la enfermedad y un pronóstico desfavorable.7
El tratamiento se basa en la resección quirúrgica, siempre que sea posible, y se completa con quimioterapia y en casos con ganglios regionales se realiza radioterapia. Según el estadiamiento The international Neuroblastoma Pathology Classification (ISPN), la supervivencia es de 5 años en los estadios del I-V en 95 %, 75-85 %, 40-70 %, -10 % y 60-90 %, respectivamente.8
Entre las masas más frecuentes del espacio presacro para realizar el diagnóstico diferencial del caso que se presenta, encontramos los tumores congénitos como el cordoma o el teratoma. El cordoma sacro comprende 50-70 % del total de las localizaciones. Se representa radiológicamnete como una masa sólida con componentes quísticos y calcificaciones amorfas. En rayos X simple se detecta por la destrucción al nivel del sacro y expansión ósea. La TAC y la RM definen más los límites de la lesión y la participación de partes blandas en el caso de la TAC. El teratoma sacrocoxígeo es el tumor presacro más frecuente en la infancia, sobre todo antes de los 2 años. Entre los tumores óseos primarios a este nivel se encuentran osteosarcoma, condrosarcoma, tumor de células gigantes, plasmocitoma u osteoma. Esta localización también es asiento de metástasis a partir de tumores de la pelvis.
REFERENCIAS BIBLIOGRÁFICAS
1. Peydro Olaya A, Llombart Bosch A, Carda Batalla C, López Guerrero JA. Electron microscopy and other ancillary techniques in the diagnosis of small round cell tumors. Semin Diagn Pathol. 2003;20(1):25-45.
2. Navarro S, Noguera R, Pellin A, López Guerrero JA, Roselló Sastre E, Cremades A, Llombart Bosch A. Atypical pleomorphic exotraosseous Ewing tumor/peripheral primitive neuroectodermal tumor with unusual phenotypic/genotypic profile. Diagn Mol Pathol. 2002;11(1):9-15.
3. Llombart Bosch A, Navarro S. Immunohistochemical detection of EWS and FLI-1 proteins in Ewing sarcoma and primitive neuroectodermal tumors: comparative analysis with CD99 (MIC-2) expresión. Appl Immunol Mol Morphol. 2001;9(3):255-60.
4. Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nature Rev Cancer. 2003;3:203-16.
5. Noguera R, Cañete A, Pellin A, Ruiz A, Tasso M, Navarro S, et al. MYCN gain and MYCN amplification in a stage 4s neuroblastoma. Cancer Genet Cytogenet. 2003;140:157-66.
6. Shimada H, Ambros I, Dehner LP. The international Neuroblastoma Pathology Classification (the Shimada system). Cancer. 1999;86:364-72.
7. Burgues Gasion O. Valor histopronóstico de la clasificación histopatológica (INPC) en el neuroblastoma infantil. Estudio comparativo con otros factores pronósticos (tesis doctoral). Universidad de Valencia; Julio 2003.
8. Navarro S. Classificacion Internationale des Neuroblastomes. Groupe d'etude Europeen du Neuroblastome. Communaute Europeenne. LNESG1. Paris; 13 de noviembre 2003.
Recibido: 16 de agosto de 2010.
Aprobado: 2 de noviembre de 2010.
Dra. Eileen de la Nuez Cobas. Facultad "Dr. Salvador Allende", Calzada del Cerro No. 1551. La Habana, Cuba. Correo electrónico: eileennuez@infomed.sld.cu

