










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La panmielosis aguda con mielofibrosis (PMAF) es un raro desorden hematológico, definido como un subtipo de leucemia aguda según la clasificación de la Organización Mundial de la Salud (OMS), de las enfermedades mieloproliferativas. Se caracteriza por un inicio abrupto con una pancitopenia en sangre periférica, escasos mieloblastos, < del 5 % y una ligera esplenomegalia.1,2 La biopsia de medula ósea muestra diferentes grados de fibrosis reticulinica y/o colágeno incremento del número de células inmaduras de las tres series desviación izquierda con algunos neutrófilos inmaduros. La presencia de normoblastos, basófila o monocitosis es rara, la serie eritroide no muestra alteraciones. Se caracteriza por una pobre respuesta y una sobrevida muy corta de alrededor de 6 meses o 1 año. Actualmente no existe una terapia estándar.3,4,5 Se presenta un paciente con el diagnóstico de una leucemia mieloide aguda no promielocítica por medulograma y inmunofenotipo con extensa mielofibrosis con esclerosis en biopsia de medula ósea, el cual recibió tratamiento con esquema 3+7 (Citosar+Rubidomicina) presentando múltiples complicaciones, así como una aplasia medular severa postquimioterapia, logrando la recuperación hematológica. Posteriormente inicia tratamiento con lenalidomida, ácido zoledronico como factor estimulante de colonias eritroides y se encuentra en remisión hematológica completa hasta marzo 2020.
Caso clínico
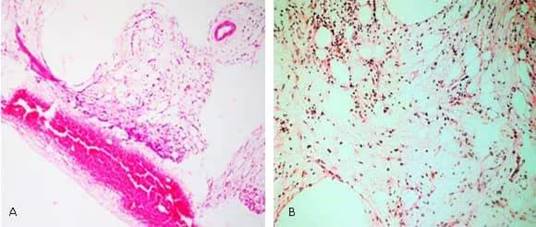
Paciente KOL, masculino, raza negra de 31 años de edad, procedente de Trinidad y Tobago, quien comienza con letargia, decaimiento, fiebre vespertina y en ocasiones gingivorragias. Por esta razón, 3 meses después acude a consulta en donde le realizan estudios y se constata pancitopenia en periferia y escasas células inmaduras. Se le realiza medulograma el cual no fue útil para diagnóstico por escaso material, así como estudios de Inmunofenotipo por citometría de flujo que reporto 30 % de células anormales con incremento de mieloblastos por los marcadores (CD34, CD13, CD17, CD117, CD38) y disminuidos en (CD11c y HLA-DR), desviación izquierda de los granulocitos. Los hallazgos consisten en una leucemia mieloide aguda sin otra especificación. Se remite a nuestro país con el diagnóstico de una leucemia mieloide aguda para tratamiento. Se realiza medulograma donde se observa escaso material, se concluye como una leucemia aguda no promielocitica. La biopsia de medula ósea (BMO) muestra una medula hipocelular global sustituida por tejido fibroconectivo correspondiente a una mielofibrosis. No se observa infiltración tumoral (fig. A). Los estudios Citogenéticos y Moleculares fueron negativos para: Duplicación interna en tándem del gen FLT3, Traslocación (8,21) (FISH), Inversión 16 o gen de fusión CBFβ-MYH11(FISH), BCR-ABL por RT-PCR, Mutación del exón 12 del gen NPM1 por RT-PCR. Cariotipo: Normal. Con estos resultados con el posible diagnóstico de Panmielosis aguda con mielofibrosis se inicia tratamiento de inducción con esquema (3+7) Citosa 100 mg/m2 + Rubidomicina 60 mg/m2. El día 11 posquimioterapia en aplasia medular severa desarrolla abdomen agudo y con el diagnóstico presuntivo de apendicitis aguda requirió tratamiento quirúrgico de urgencia por posible apendicitis aguda y resultó ser un gran hematoma inter asas. Evolutivamente mejora el cuadro doloroso abdominal, pero presenta hipopotasemia severa con distensión abdominal, enterorragia, se realizó TAC de abdomen observándose masa heterogénea hacia el íleo terminal de 95 x 57 mm, que parece corresponder con hematoma. Asociado a esto presentó sepsis polimicrobiana con el uso de múltiples antibióticos de amplio espectro, hemocomponentes y factores estimulantes de colonias y desarrolló una aplasia medular prolongada, por lo que se decide realizar medulograma y BMO evolutivos el día 28 posquimioterapia debido a la persistencia de la pancitopenia.
En el medulograma no se obtuvo material útil para diagnóstico y en la BMO se observa una médula ósea con hipocelularidad marcada a expensas de los tres sistemas, acompañado de mielofibrosis y esclerosis severa correspondiente a una mielofibrosis secundaria severa con abundante pigmento hemosiderínico (fig. B). Evolutivamente mejoría clínica y hematológica, no evidencias de células inmaduras en periferia y medula ósea, Se realiza estudio HLA (histocompatibilidad) para posible trasplante alogénico y se inicia tratamiento de consolidación con Lenalidomida 10 mg diarios y Zometa 4 mg con frecuencia mensual, así como apoyo con factores estimulantes de colonias (Eritropoyetina y Leukocim). Se egresa luego de 73 días de hospitalización y en la actualidad mantiene tratamiento impuesto con recuperación hematológica, y seguimiento en consulta externa de hematología.
Discusión
La PMAF según la clasificación de la OMS se ha definido como un subtipo raro de leucemia mieloide aguda de las enfermedades mieloproliferativas que representa 2 % de estas.1,2,3,4,5 Este desorden se caracteriza por una proliferación panmieloide aguda con una fibrosis a nivel de la medula ósea. Clínicamente se caracteriza por una ligera esplenomegalia y una pancitopenia en sangre periférica, escasos mieloblastos < de 5 %. En la biopsia se médula ósea se pueden encontrar diferentes grados de fibrosis reticulinica o colágena desviación izquierda con algunos neutrófilos inmaduros.5,6 Existe un incremento de las células inmaduras hematopoyéticas con focos de escasos blastos. La respuesta es pobre con la quimioterapia y está asociada a una pobre sobrevida.4,6 Un desafío de la PAMF resulta el diagnóstico diferencial con síndromes mielodisplásicos asociados a mielofibrosis, leucemia aguda megacarioblástica con mielofibrosis que representa de 3-5 % de la leucemia mieloide aguda, la LMA con displasia multilinage y mielofibrosis y la mielofibrosis primaria, así como la exposición a agentes mielo tóxicos7,8,9 por lo que el diagnostico en la mayoría de los casos es extremadamente difícil.
En la actualidad, no existe un esquema de quimioterapia estándar para el tratamiento de esta entidad se proponen esquemas de inducción 3+7 (Citosar 100 mg/ m2 + Rubidomicina 60 mg/ m2) seguidos de un trasplante de progenitores hematopoyéticos alogénico.10,11,12,13 Los reportes acerca del trasplante alogénico en esta entidad no muestran resultados alentadores como lo muestra el estudio japonés de 40 pacientes sometidos a trasplante alogénico, en un periodo de 10 años con una sobrevida global a los 3 años de 24 %.10 Otros autores plantean luego del tratamiento de inducción iniciar, ácido zoledronico bifosfonato de tercera generación con buena respuesta clínica y desaparición de la fibrosis y los blastos asociado a lenalidomida y factor estimulantes de colonias granulocíticas y Eritroides. Los bifosfonatos son agentes útiles en la osteoporosis, Enfermedad de Paget, mieloma múltiple e hipercalcemia, también se ha probado con éxito en la mielofibrosis idiopática.13
La PAMF no es una causa común de pancitopenia; sin embargo, debe ser considerada en adultos jóvenes donde no existe una causa infecciosa o toxica demostrada con pancitopenia y escasos blastos en periferia muchas veces sin esplenomegalia. En el medulograma la aspiración es blanca y la biopsia de médula ósea muestra diferentes grados de fibrosis. La correlación clínica con los elementos anatomopatológicos (biopsia de médula ósea, inmunohistoquímica) y estudio inmunofenotipo expresando la positividad de los blastos para CD34, CD13, CD33, así como CD117, negativo para la mieloperoxidasa y línea megacariocitica, constituyen el Gold estándar en el diagnóstico de esta entidad. El estudio citogenético permite el diagnóstico diferencial fundamentalmente con los síndromes mielodisplásicos y la leucemia megacarioblástica. La PAMF es una entidad muy infrecuente con una sobrevida muy corta y un pronóstico reservado a pesar de los esfuerzos terapéuticos que en la actualidad se llevan a cabo.

