










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduccio´n
El pénfigo pertenece a un grupo de enfermedades mucocutánea vesícula-erosivas de origen autoinmune, por tanto los autoanticuerpos van dirigidos contra los desmosomas de los queratinocitos, lo que ocasiona acantólisis y la formación de vesículas intraepiteliales.1 Las lesiones orales se han asociado solo al pénfigo vulgar (PV) y al pénfigo paraneoplásico (PNP). EL PNP fue descrito como una entidad individualizada del resto de los pénfigos en el año 1990 en un estudio realizado en 5 pacientes que presentaban erosiones mucocutánea extensas y una neoplasia subyacente.2
El pénfigo paraneoplásico es una rara enfermedad autoinmune ampollosa que típicamente se manifiesta con una estomatitis hemorrágica y erosiones extensas de la mucosa, asociada a una neoplasia obvia u oculta, dos tercios de los pacientes tienen un proceso tumoral ya diagnosticado en el momento de aparición de los primeros signos, generalmente es un proceso linfoproliferativo, pero en más del 30 del PNP es la primera manifestación del proceso subyacente, por lo que es necesario un estudio completo.
La etiopatogenia del PNP no se conoce por completo.3,4 Se asume que las lesiones cutáneas son causadas por una respuesta autoinmune generada por anticuerpos contra antígenos tumorales que reaccionan de forma cruzada con antígenos epiteliales.
No hay consenso con respecto a los criterios de diagnóstico del PNP, pero se sospecha ante signos como: dolor e inflamación de la mucosa oral, lesiones vesículo-ulcerativas con signo de Nikolsky positivos, una erupción cutánea polimorfa con correspondencias histológicas que a menudo muestran cambios liquenoide o acantolítica.
Estudios de inmunofluorescencia directa e indirecta son los exámenes ideales para el diagnóstico, que revelan anticuerpos séricos anti-DSG, desmoplaquina I y II, envoplaquina, periplaquina, antígeno prefijoide bulloso tipo 1 y anticuerpos de plectina en la unión intercelular y de la membrana basal.
La tasa de mortalidad oscila entre el 75 y el 90 % y la principal causa de muerte en estos pacientes es la insuficiencia respiratoria.5,6 El diagnóstico precoz y el inicio temprano del tratamiento son de suma importancia. Presentamos un paciente, con tumor renal respectivamente, con compromiso mucocutánea, cuya clínica y hallazgos inmunopatológicos corresponden al diagnóstico de pénfigo paraneoplásico.1,3
Presentación de caso
Se presenta un paciente de sexo masculino de 47 años de edad con antecedentes de asma bronquial desde la infancia, contralado con salbutamol (spray), con hipertensión arterial (HTA) desde hace tres años, controlado con enalapril 20 mg, media tableta cada 12 h, hidroclorotiazida de 25 mg una tableta diaria; además de catarata y reflujo gastroesofágico por hernia hiatal; niega antecedentes familiares de interés.
Motiva su ingreso por lesiones erosivas de la mucosa oral, dolorosas con costras hemorrágicas y saliveo constante con afectación de carrillos, lengua, encías y también con lesiones de mucosa conjuntival y nasal. Las lesiones de mucosa oral se presentaron desde el año 2018 y nunca mejoraron con los tratamientos tópicos utilizados.
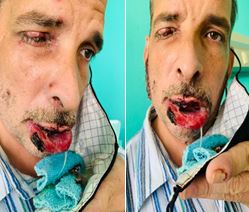
El examen físico reveló una cavidad oral difícil de explorar, se apreció edema labial y de la mucosa con abundante sialorrea, áreas denudadas de toda la mucosa con restos de esfacelos blanquecinos en mucosa labial y de los carrillos; erosiones del bermellón a todo lo largo y costras hemorrágicas y necróticas en el labio superior, área del bigote y en el vestíbulo nasal (fig. 1).
En el examen cutáneo las lesiones eritematocostrosa y hemorrágicas en las placas fueron desde 1 cm de diámetro hasta 7 cm, que aparecen distribuidas en el tronco anterior y la espalda (fig. 2).
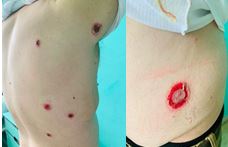
Fig. 2 Examen cutáneo en el tronco anterior y espalda con lesiones eritematocostrosa y hemorrágicas.
En el dedo índice de la mano izquierda y el primer dedo del pie derecho se apreció un edema, un eritema con costras de la matriz ungueal y los repliegues laterales, donde existió ampolla referida por el paciente y una onicodistrofía (fig. 3).
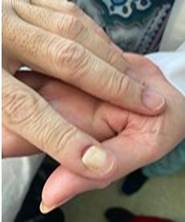
Fig. 3 Eritema con costras de la matriz ungueal y repliegues laterales donde existió ampolla. Signo de Nikolsky marginal y a distancia negativo.
En las pruebas de laboratorio se observaron: leucocitosis (21,41 x 109/L) y neutrofilia (85,1 %), glucemia de 9,76 mmol/L, TGP 200 U/L y TGO 150 U/L, triglicéridos 2,8 mmol/L, cituria con abundantes bacterias, el examen bacteriológico de lesiones cutáneas y de la mucosa conjuntival con Staphylococcus aureus, el examen micológico negativo, y PCA 0,751 ng/mL.
La biopsia de piel (B21-3611): la biopsia cutánea reveló acantólisis suprabasal y queratinocitos necróticos. Se informó como resultado pénfigo vulgar. IFD con depósitos de IgG y C3 alrededor de los queratinocitos (fig. 4).

En cuanto al TAC de tórax y el abdomen se reportaron bandas de atelectasias subsegmentarias vs tractos de fibrosis en bases pulmonares (segmentos posteriores), sin derrame pleural y pericárdico. Granuloma calcificado en LID, cambios osteodegenerativos con protrusión discal de base ancha y compresión del saco dural. En el radiodiagnóstico (RD) se observó una masa heterogénea hacia el polo superior del RD, por el cual aparece tener un centro hiperdenso rodeado de un halo hipodenso, se observó; además, signo del pilo, demostrándose dependencia del riñón.
En la AngioTAC se informó la existencia de una masa tumoral con realce en la captación del contraste, el cual se ubicó hacia el polo superior del RD. Dicha lesión presentó un centro más hipodenso en relación con una necrobiosis asociada que provoca una amputación parcial del cáliz superior, conjuntamente con lo anterior es evidente la participación en su aferencia de la suprarrenal inferior, la cual irriga el polo superior del propio tumor (fig. 5).
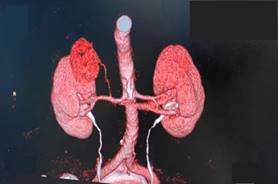
Se realiza una interconsulta con urología que propone embolización del tumor.
Tratamiento en la hospitalización: se utilizaron bolos de metilprednisolona en número de 3 diarios (500 mg/día), después se utilizó 60 mg de prednisolona oral diario, la ceftriaxona 1 g EV cada 12 h por 7 días y después ciprofloxacina 2 (bb) EV cada 12 h, realizar varias veces al día buches mucolíticos, fomentos en áreas erosivas de la piel y cremas esteroideas, más cremas antibióticas, después de los fomentos. El paciente se mantiene hace 1 año sin actividad cutánea mucosa y con tratamiento de prednisolona 1 tableta de 20 mg diarios; además, mantiene un seguimiento por la Consulta de Urología y por la de Dermatología cada 3 meses.
Discusio´n
El pénfigo paraneoplásico es una enfermedad ampollosa autoinmune que se presenta en el contexto de una neoplasia oculta o previamente diagnosticada, descripta por Anhalt en 1990 en un estudio realizado en 5 pacientes que presentaban erosiones mucocutánea extensas y una neoplasia subyacente.
En el año 2001, se propuso el término de síndrome multiorgánico autoinmune paraneoplásico (PAMS) debido al compromiso de múltiples órganos; además del cutáneo por presentar una patogenia que no se limita a la presencia de autoanticuerpos contra el complejo antigénico conocido.1,3
Este cambio de nombre es controvertido por varios motivos, en primer lugar, el nombre original pénfigo paraneoplásico rinde homenaje al Dr. Grant Anhalt que describió esta entidad, acuñó el término, y ha llevado muchos años investigando en este campo.5,6 En segundo lugar, a pesar de que la nueva denominación pretende ser descriptiva, puede crear mayor confusión entre la comunidad científica, ya que pueden haber otros cuadros que también encajen dentro de ese nombre.6,7,8,9
Como su nombre indica, la presencia de neoplasia es constante en el PPN. El tipo de neoplasia asociado depende del grupo de edad afectado. En los adultos con PPN en el momento del diagnóstico, la neoplasia subyacente se conoce en dos tercios de los casos.3
Las neoplasias que se observan más frecuentemente asociadas en adultos son: linfomas no hodgkinianos, 42 %; leucemia linfática crónica, 29 %; enfermedad de Castleman, 10 %; macroglobulinemia de Waldeström, 6 %; timomas, 6 %, y sarcomas, 6 %. Por lo que respecta al PPN por debajo de los 18 años, la neoplasia subyacente suele desconocerse en el momento del diagnóstico en un 70-80 % de los casos descritos.5,6,9
La mayoría de estos casos están asociados a la enfermedad de Castleman, que representaría el 70 % de las neoplasias, y es excepcional la asociación al linfoma no hodgkinianos o leucemia linfática crónica.
En el paciente en estudio se presenta una neoplasia de células clara de riñón derecho que se extiende a la suprarrenal y a la vena cava, cuyo diagnóstico fue realizado en el ingreso y una estomatitis severa, dolorosa con toma de la mucosa conjuntival y nasal; además de una afectación cutánea con placas costrosas, hemorrágicas, erosivas, localizadas en la cara, en el tronco anterior y posterior y la espalda, por lo que resultó motivo de interés para nuestra investigación.5,6,8,9
El PPN se presenta con mayor frecuencia entre los 45 y 70 años, aunque el rango de edad varía entre los 7 y 83 años. El mecanismo fisiopatológico, por el cual se producen las lesiones mucocutánea en el PPN no se conocen con exactitud.1,3,5,6 Intervienen tanto la inmunidad celular como la humoral.
Un hallazgo constante, y de primer signo de la enfermedad es el compromiso de la mucosa oral. Es característica la presencia de erosiones y ulceraciones dolorosas que afectan la mucosa y se extienden hasta el bermellón de los labios de muy difícil manejo y refractarias al tratamiento.
Otras mucosas como la conjuntival, la esofágica y la anogenital también pueden estar comprometidas.3,4).Las lesiones cutáneas son muy variadas de ampollas fláccidas o tensas, con o sin erosiones y/o lesiones de tipo eritema multiforme, liquen plano o injerto contra huésped. Este polimorfismo lesionar puede estar presente en un mismo paciente en un determinado momento o a lo largo de la evolución de la enfermedad. El árbol traqueobronquial puede estar directamente comprometido, y puede producir un fallo respiratorio por la presencia de bronquiolitis obliterante debido a los autoanticuerpos contra las plaquinas del epitelio bronquial, que serían los responsables de los cambios acantolíticos en este.
Según lo anteriormente expuesto esta afección se presenta en el 30 % de los casos y puede desarrollarse al mes o hasta un año posterior al diagnóstico. Debemos sospechar ante la presencia de la dificultad respiratoria progresiva.4,5,6,9 Dentro de los diagnósticos diferenciales debemos tener en cuenta las enfermedades que cursen con estomatitis refractarias, entre ellos el eritema multiforme, el síndrome de Steven Johnson, la necrolisis epidérmica tóxica, el pénfigo vulgar, el prefijoide ampollosa, el liquen plano, la dermatitis por radiación e infecciones nicóticas o herpéticas.
El tratamiento de esta enfermedad puede dividirse en dos categorías, por un lado el manejo de la neoplasia y por otro el fenómeno autoinmune.6,7 La mayoría de los pacientes con afecciones benignas mejoran o se curan luego de la extirpación quirúrgica.
En el caso de una neoplasia maligna, no existe una terapéutica eficaz, a pesar de la extirpación quirúrgica de esta y/o del uso de la quimioterapia, esto no impide la progresión de la enfermedad. En aquellos casos que se presentan con bronquiolitis obliterante el pronóstico es reservado.
Para el PPN los corticoides a altas dosis son el tratamiento de elección. El uso concomitante de drogas inmunosupresoras reduciría la dosis de esteroide, lo que limitaría los efectos adversos. La combinación más frecuentemente utilizada es la meprednisona, 1-2 mg/kg/d y ciclosporina A, 5 mg/kg/d. Sin embargo, no se han obtenido resultados favorables. Tampoco el uso de plasmaféresis, gamma globulina endovenosa, dapsona, ni las sales de oro ha demostrado ser efectivos.
Se han publicado resultados prometedores con el uso del rituximab, anticuerpo monoclonal anti CD20, en los casos de PPN y linfoma de células B. El pénfigo paraneoplásico presenta una tasa de mortalidad entre el 75 y 90 %. Las causas de muerte suelen ser las complicaciones de la enfermedad y/o del tratamiento como: sepsis, falla multiorgánico, hemorragias gastrointestinales o fallo respiratorio, producto de la bronquiolitis obliterante.4,8,9
Se concluye que el PPN es un desorden con características clínicas e histopatológicas bien definidas asociado a una neoplasia linfoproliferativa benigna o maligna. En el caso presentado, dado el compromiso cutáneomucoso en asociación con el tumor renal se demuestra la complejidad y el polimorfismo de la enfermedad inmunoampollosa. Por tanto, es esencial reconocer la importancia de la correlación de los hallazgos clinicopatológico e inmunológicos en el PPN, así como la posibilidad de que se produzcan presentaciones que urjan la pronta confirmación y evaluación inmunopatológica de una neoplasia oculta, como en el caso estudiado que presentó un tumor renal que con frecuencia representa el 16 %, asociada al desarrollo de un PPN. La respuesta al tratamiento depende del tipo de neoplasia y de las complicaciones.

