










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr v.69 n.2 Ciudad de la Habana Mayo-ago. 1997
Síndrome frágil X: correlación clínica, citogenética y molecular en una familia
Dra. Aracely Lantigua Cruz,1 Dra. Ileana Ravelo Amargos,2 y Dra. Dicky Halley3RESUMEN
Estudios recientes han permitido caracterizar molecularmente el defecto genético o mutación FMR-1 del síndrome frágil X. Esta mutación consiste en variaciones de repeticiones del triplete CGG del gen, que varían del rango normal a la mutación completa y pasan por rangos intermedios o premutaciones. Debido a que el individuo afectado además expresa un porcentaje variable de sitio frágil Xq27.3 (FRAXA) en su cariotipo, en este trabajo se presenta una correlación clínica y citogenética con la caracterización molecular del gen FMR-1 realizada por 2 métodos directos: Southern blot y reacción en cadena de la polimerasa (PCR) en 5 varones afectados y 5 mujeres portadoras, a propósito del diagnóstico prenatal en una de ellas. El análisis de las caracterizaciones clínicas, físicas y neuropsicológicas previamente delineadas, llevaron a la conclusión de que éstas, así como la frecuencia en porcentaje de FRAXA, se correlacionan en los varones afectados con la elongación que experimenta la mutación al trasmitirse de una generación a la siguiente a través de mujeres portadoras, en quienes por otra parte, estas características son heterogéneas y susceptibles a sobrelapamiento tanto por efecto genómico como por factores ambientales psicofamiliares.Descriptores DeCs: SÍNDROME DE FRAGILIDAD DEL CROMOSOMA X/genética; GENÉTICA BIOQUÍMICA; SOUTHERN BLOTTING/métodos; REACCIÓN EN CADENA POR POLIMERASA/métodos; CITOGENÉTICA.
Pocas enfermedades genéticas han despertado tanto interés como el síndrome Frágil X.
Desde que en 1943 Martin y Bell1 reportaron claras evidencias de herencia recesiva ligada al cromosoma X en una familia en la que los varones presentaban retraso mental, pasando por el reporte de Lubs en 19692 acerca de la relación entre el sitio frágil en el extremo distal de los brazos largos del cromosoma X, hasta la caracterización molecular del gen FMR-1,3 han transcurrido apenas 48 años.
Con los conocimientos actuales comienza una nueva etapa de investigaciones destinada a dilucidar el defecto básico que media entre el conocimiento molecular de esta segunda causa genética de retraso mental (RM).
Nuestro propósito con este trabajo ha sido el de analizar la correlación existente entre las manifestaciones clínicas previamente delineadas, porcentaje de fragilidad Xq27.3 y la caracterización molecular del gen FMR-1, en 5 varones afectados y 4 mujeres portadoras obligadas de 3 generaciones de una familia registrada en el servicio de Genética Clínica del Hospital Pediátrico Docente "Juan Manuel Márquez".
MÉTODOS
Estudios clínicos: A solicitud de diagnóstico prenatal de una mujer portadora obligada, previamente asesorada genéticamente, se procedió al examen clínico de los 5 varones registrados como afectados en las 3 generaciones componentes de la familia, así como de las 5 mujeres, y se seleccionaron los signos dismórficos y alteraciones del tejido conectivo, así como características neuropsicológicas que han sido previamente delineadas y atribuibles por su frecuencia, en pacientes no emparentados, con el defecto genético4,5 y que se enumeran a continuación:Características clínicas físicas:
- Facie no específica.
- Cara alargada.
- Orejas grandes y prominentes.
- Mentón grande.
- Paladar alto.
- Macroorquidismo.
- Hiperlaxitud articular.
- Pectus excavatum o carinatum.
- Coeficiente de inteligencia (CI) superior a 70.
- CI entre 70 y 50.
- CI inferior a 50.
- Lenguaje reiterativo.
- Hiperactividad.
- Evitación con la mirada.
- Epilepsia.
Volumen testicular (mL) = p /6 x l x a
donde: l = longitud del testículo y
a = ancho del testículo.
Se consideró macroorquidismo cuando el volumen testicular excedió los 25 mL en el adulto y los 2 mL en los prepuberales.6
La severidad de RM de los miembros de la familia se obtuvo mediante las pruebas psicométricas realizadas en el Centro de Orientación y Diagnóstico y se añadió una valoración cualitativa al tener en cuenta las esferas que afectan de manera peculiar a los pacientes según estudios neuropsicológicos reportados en la literatura médica7 y se valoró el desarrollo del lenguaje, la comunicación verbal y extraverbal, relaciones sociales, habilidades adquiridas y conducta general.
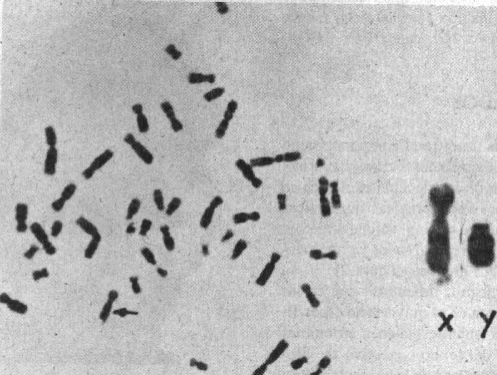
Estudios citogenéticos
Se tomó sangre periférica heparinizada y se cultivó pocas horas después de obtenida ésta en medio 199 (folato deficiente) y sin suplemento exógeno y según la microtécnica de cultivo de muestra de laboratorio (Lantigua Cruz A, Viñas Portillo C, Delgado Oceguera N, Monte E del: Microtécnica de cultivo de sangre periférica utilizando plasma del propio paciente como suplemento, ANIR, 1987). Previa incubación a 37 oC y 72 horas se procedió a la preparación final según técnicas convencionales de rutina.Las láminas se trataron con tripsina y coloreadas con giemsa para la obtención de bandas G (GTG).
Una vez excluidas otras aberraciones cromosómicas se procedió al análsis de 100 metafases para la detección del sitio frágil Xq27.3.
Caracterización molecular
Se obtuvieron 10 mL de sangre de 9 individuos utilizando como anticoagulante etilendiaminotetracetato (EDTA), y una muestra de vellosidad coriónica por biopsia a la portadora embarazada (II-3 en el árbol genealógico de la figura 1).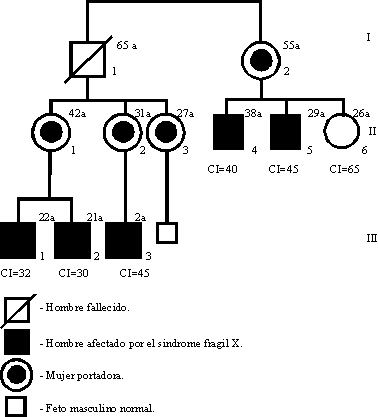
Las muestras se enviaron al departamento de genética clínica de la Universidad de Erasmus en Rotterdan, Holanda, donde se extrajo el ácido desoxinucleótico (ADN) por métodos convencionales y se procesaron las muestras con 2 métodos directos para la caracterización del gen FMR-1:
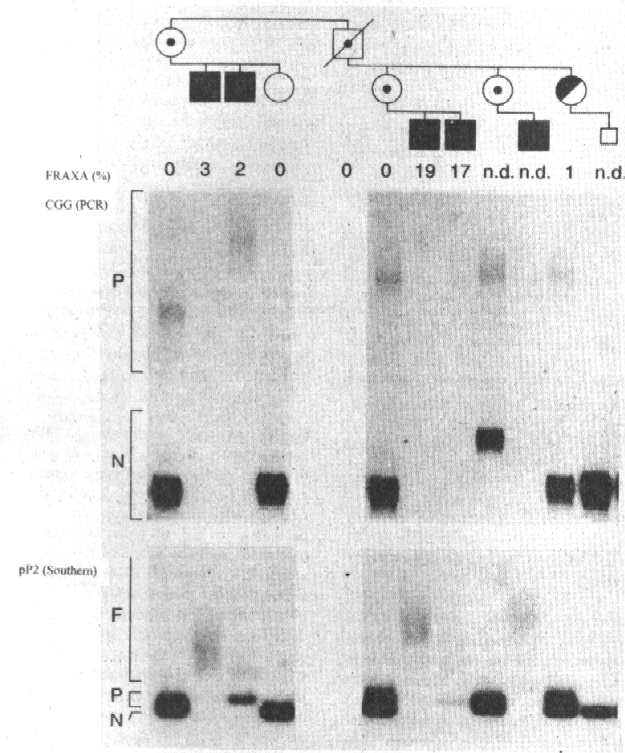
- Southern blot previa digestión del ADN con la endonucleasa de restricción Hind III y posterior hibridación con el probe pP2, y se denominó N al alelo normal e I a las inserciones CGG en rangos de premutaciones (P) o de mutaciones completas (F) o mosaicismo de ambas (P + F).
- Reacción en cadena de la polimerasa (PCR) para precisar el número de repeticiones CGG del gen FMR-1, en la cual se consideró alelo normal de 0 a 50 repeticiones; premutación de 50 a 200 y cuando hubo ausencia de señal, mutación completa con rangos superiores a 200 repeticiones CGG.
RESULTADOS
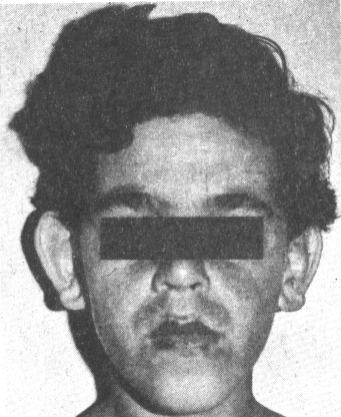
La tabla muestra el comportamiento de las manifestaciones físicas y neurosicológicas según la selección explicada en métodos. Las características dismórficas más constantes en los varones fueron: cara alargada, orejas grandes y prominentes, mentón grande (figura 2) y el macroorquidismo, mientras que en las mujeres fueron en su mayoría no específicas. De las manifestaciones atribuibles a defectos del tejido conectivo se destaca la hiperlaxitud de las articulaciones. Los rasgos neuropsicológicos y el RM inferior a 50 fueron evidentes en los varones y no específicos en las mujeres de esta familia. El 80 % de los varones presentan epilepsia.| | |||||||
| | | ||||||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | |||
En la figura 3 se muestran las características del sitio frágil Xq27.3. Los estudios moleculares y citogenético de la portadora II-2 y de su hijo afectado III-3 (figura 1) se realizaron simultáneamente. El hombre I-1 fue estudiado citogenéticamente antes de su fallecimiento. Exceptuando a la mujer II-3 con 1 % de sitio frágil, el resto de las mujeres analizadas así como el hombre I-1 no mostraron sitio frágil Xq27.3. Los varones II-4 y II-5 tuvieron 3 y 2 % respectivamente y los valores más altos fueron vistos en III-1, III-2 y III-3 con frecuencias de 19,17 y 15 % de sitio FRAXA.
La caracterización molecular aparece en la figura 4, en la que se destaca en el diagnóstico prenatal de transmisión del cromosoma X normal de la portadora II-3 a su feto.
La correlación clínica citogenética y molecular se expresa en la figura 5 en la que se evidencia un incremento de severidad de las manifestaciones clínicas, así como de porcentaje de fragilidad en correspondencia con el paso de la mutación en las generaciones estudiadas.

DISCUSIÓN
El 80 % de los varones pospuberales presentan cara alargada, orejas prominentes y macroorquidismo;8 en nuestros pacientes éstos se presentaron en el 100 % excepto II-5 que no presenta cara alargada.Los defectos propios del tejido conectivo como paladar alto y pectus excavatum o carinatum se observaron solamente en los varones afectados de la tercera generación, hecho éste no correlacionado con la edad y sí probablemente con la magnitud de la mutación completa. La hiperlaxitud articular es mucho más generalizada para todos los valores afectados y también en dependencia de la edad.5
Las manifestaciones neuropsicológicas estudiadas son los elementos más significativos del síndrome. El RM aunque con CI inferior a 50, es más severo en los varones de la tercera generación, y se correlaciona con el porcentaje de fragilidad incrementada en estos pacientes, que por otra parte, como se ha reportado9 se hace evidente cuando el número de repeticiones CGG es superior a 200, debido posiblemente a la baja disponibilidad de citidina en medios deficientes de folato que interfieren la síntesis in vitro de las expansiones CGG.10 De ser así, predictivamente la familia estudiada, la expansión de la mutación completa sería mucho mayor en los varones de la tercera generación que en los de la segunda.
Aunque la fisiopatología del síndrome no se ha confirmado, se sabe que cuando la elongación de CGG de la mutación es muy grande las regiones CpG adyacentes se hipermitilan y afectan a la transcripción del gen y a su vez la síntesis de la proteína que ésta codifica,11 lo que pudiera ser la ausencia de esta proteína la responsable de las manifestaciones neuropsicológicas descritas.11
A su vez el incremento de repeticiones CGG depende del tamaño de la premutación y el sexo del portador. Las premutaciones que transmite un hombre portador sano no varían al ser transmitidas a sus hijos, pero éstas se pueden incrementar notablemente al ser transmitidas a sus nietos.12
En esta familia esto sería otra explicación de la severidad clínica en los pacientes de la tercera generación respecto a la segunda, pues la mujer I-2 presenta una premutación de 80 repeticiones mientras que en las portadoras obligadas de la generación III la premutación es de 90 repeticiones para incrementarse durante la ovogénesis el riesgo de pasar a una mutación completa mucho mayor que la de los varones de la generación II.13
La epilepsia ha sido reportada y estudiada por otros autores al describir las anormalidades neurológicas del síndrome (Rodríguez H, Lantigua A, Guerra D. Síndrome frágil X en 35 varones con retraso mental inespecífico [trabajo de terminación de residencia de Genética Clínica] Centro Nacional de Genética Médica, ISCM-H, 1987). Se ha reportado correlación entre las convulsiones y el porcentaje de sitio frágil Xq27.3.11 En esta familia sólo un varón de la III generación no ha presentado epilepsia, lo que sugiere que este síntoma tal vez no dependa solamente de la mutación.
A pesar de que el hombre I-1 no pudo ser caracterizado molecularmente, el hecho de tener una hermana y todas sus hijas portadoras evidencian que él es un hombre transmisor sano. El 20 % de los varones portadores son fenotípicamente también normales, sus hijas son portadores y fenotípicamente también normales, mientras que sus nietos tendrán alto riesgo (40 %) de presentar el síndrome.14
Aunque no es el objetivo de este trabajo es importante señalar que la caracterización molecular del gen es de extraordinario valor para el diagnóstico prenatal, sobre todo si, como en este caso el feto varón hereda el cromosoma X normal de la madre.
Podemos concluir con que las manifestaciones físicas y neuropsicológicas en los varones afectados son más constantes y se pueden relacionar con la expresión de diferentes elongaciones CGG en la mutación completa del gen FMR-1.
En esta familia se puede correlacionar entre expresión de FRAXA y la elongación de la mutación completa FMR-1 al apoyar la idea de correspondencia entre ésta y la baja disponibilidad de citidina por la deficiencia de folato del medio de cultivo utilizado, que interfiere en el completamiento de la síntesis, in vitro de la expansión CGG y que se evidencia microscópicamente como un gap o ruptura al nivel de Xq27.3. cuando existen más de 200 repeticiones.
El fenotipo de las portadoras en nuestro estudio no permite establecer una correlación específica. El perfil neuropsicológico es susceptible de sobrelapamiento con otras posibles alteraciones genómicas o por factores ambientales psicofamiliares. La hija (II-6) de la mujer I-2 comparten fenotipos similares y genotipos diferentes, mientras que I-2 es heterocigótica, II-6 presenta 2 cromosomas X con el rango normal de repeticiones CGG.
AGRADECIMIENTOS
A los profesores H. Galjaard del Departamento de Genética Clínica de la Universidad Erasmus en Rotterdan, Holanda y Luis Heredero del Centro de Genética Médica del Instituto de Ciencias Médicas de La Habana, por su apoyo en el estudio molecular y envío de las muestras.SUMMARY
Recent studies have allowed to characterize molecularly the genetic defect or FMR-1 mutation of the fragile X syndrome. This mutation consists in variations of repetitions of the gene's CGG triplet that came from the normal range to the complete mutation, passing through intermediate ranges or premutations. Due to the fact that the affected individual also shows a variable percentage of Xq27.3 fragile site (FRAXA) in his karyotype, it is presented in this paper a clinical and cytogenetic correlation with the molecular characterization of the FMR-1 gen carried out by 2 direct methods: Southern Blot and polymerase chain reaction (PCR) in 5 affected males and 5 female carriers, one of them with prenatal diagnosis. The analysis of the clinical, physical and neuropsychological characterizations previously delineated, led to the conclusion that these, as well as the FRAXA frequency in percentage, are correlated among the affected males with the elongation the mutation suffers on being transmitted from one generation to another through female carriers, in whom these characteristics are heterogeneous and susceptible to overlapping as a result of genomic effect and of environmental psychofamilial factors.Subject headings: FRAGILE X SYNDROME/genetics; GENETICS, BIOCHE-MICAL; BLOTTING SOUTHERN/methods; POLYMERASE CHAIN REACTION/methods; CYTOGENETICS.
REFERENCIAS BIBLIOGRÁFICAS
- Martin JP, Bell JA. A pedigree of mental defect showing sexlinkage. J Neurol Neuronog Psychiatr 1943;6:154-7.
- Lubs HA. A marker X chromosome. Am J Hum Genet 1969;21:231-44.
- Verkerk AJMH, Graaff E de, De Boulle K Eichler EE, Konecki DS, Reyniers E. et al. Identification of a gene (FMR-1) containing a CCG repeat coincident with a fragile X break point cluster region exhibing lengh variation in fragile X syndrome. Cell 1991;65:905-14.
- Rodríguez Guas H, Lantigua Cruz A. Síndrome frágil X, reporte de 14 pacientes. Rev Cubana Pediatr 1989;61:21-35.
- Chudley AE, Hogerman RJ. Medical progress Fragile X syndrome. J Pediatr 1987;110:821-31.
- Prader A. Testicular size: assessment and clinical importance. En: Sutherland GR, Hecht F. Fragile sites on human cromosomes. Oxford monographs on medical genetic, Oxford:1985:280.
- Reiss AL, Freund L. Behavional phenotype of fragile X syndrome DSM-III R austistic behavior in male children. Am J Med Genet 1992;43:35-46.
- Opitz JM, Sutherland GR. Conference report: International Workshop on the Fragile X and X-Linked Mental Retardation. Am J Med Genet 1984;17:5-94.
- De Vries BBA, Wiegers AM, Graaff E de, Verkerk AJMH, van Hemel JO, Hally DJJ. et al. Mental status and fragile X expression in relation to FMR-1 gen mutation. Eur J Hum Genet 1993;1:72-9.
- Sutherland GR, Baker E, Fratim A. Excess thymidine induces folate sensitive fragile sites. Am J Med Genet 1985;22:433-43.
- Pieretti M, Zhang F, Fu YH, Warren ST, Oostra BA, Caskey CT, et al. Absence of expression of the FMR-1 gene in fragile X syndrome Cell 1991;66:817-22.
- Mandel JL. Hagerman R, Froster U, Brown, WT. Jenkins EC, Jacobs P, et al. Fifth International Workshop on the fragile X and X-linked mental retardation. Am J Med Genet 1992;43:5-27.
- Fu YH, Kuhl DPA, Pizzulti A, Pieretti M, Richards S, Verkerk AJHM, et al: Fragile X sites: a polymorphic and highly mutable CGG repeat in the FMR-1 gene. Cell 1991;67:1047-9.
- Sherman SL, Jacobs PA, Morton NE, Froster-Iskenius U, Howard Peebles PN, Nielsen KB, et al. Further segregation of the fragile X syndrome with special reference to transmitting males. Hum Genet 1985;69:289-99.
Dra. Aracely Lantigua Cruz. Departamento de Genética Clínica. Centro Nacional de Genética Médica, ISCM-H, Calle 146 y Avenida 31, Cubanacán, municipio Playa, Ciudad de La Habana, Cuba.
1 Doctora en Ciencias Médicas. Especialista de II Grado en Genética Clínica. Profesora Titular. Jefa del Departamento de Genética Clínica, Centro Nacional de Genética Médica.
2 Especialista de I Grado en Genética Clínica.
3 Investigadora del Departamento de Genética Clínica, Universidad de Erasmus.

