










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr v.70 n.1 Ciudad de la Habana ene.-mar. 1998
Reporte de casos
Hospital Pediátrico Docente «William Soler», Ciudad de La HabanaEstudio clínico, genético y molecular en un paciente con atrofia muscular espinal
Dra. Ibis Menéndez Alejo,1 Dra. Concepción Hernández Chico 2 y Dra. Fara Cepero Noriega 3RESUMEN
Las atrofias musculares espinales (AME) infantiles son enfermedades neuromusculares hereditarias caracterizadas por la degeneración de las motoneuronas alfa del asta anterior de la médula espinal. La enfermedad de Werdnig-Hoffmann, AME tipo I, es la forma más grave, se transmite como un carácter autosómico recesivo y los afectados suelen fallecer durante el primer año de vida por fallo respiratorio. En este trabajo se presenta una familia cubana con 2 hijos con AME tipo I, en la cual el estudio molecular en uno de ellos permitió identificar los 2 cromosomas parentales asociados con ésta. Se encontró además en el paciente una deleción de ambas copias del gen SMN (exón 8) y del gen NAIP (exón 5). Los hallazgos ilustran la utilidad de estos estudios, con vistas a posibilitar el diagnóstico prenatal de la enfermedad.Descriptores DeCS: ENFERMEDAD DE WERNIG-HOFFMANN; EXONAS/genética; GENETICA BIOQUIMICA; CUBA.
La degeneración de las motoneuronas del asta anterior de la médula producen cuadros clínicos conocidos como atrofias musculares espinales (AME), caracterizados fundamentalmente por hipotonía, arreflexia y atrofia muscular, proximales y simétricas.1,2 En edades pediátricas se conocen 3 formas clínicas de ella: la enfermedad de Werdnig-Hoffmann tipo I, que es la forma más grave; la enfermedad de Werdnig-Hoffmann tipo II y la enfermedad de Kugelberg Welander (tipo III), que es la forma más benigna y en la cual los pacientes afectados pueden llegar a caminar, aunque con dificultad.3-5 Todas ellas se transmiten según un patrón de herencia autosómica recesiva y están ligadas genéticamente al brazo largo del cromosoma 5, en la región q11.2-q13.3,6-9 lo que sugiere que son producidas por mutaciones diferentes en el mismo locus.6-12 Este trabajo se realizó en colaboración con la Unidad de Genética Molecular del Hospital "Ramón y Cajal" y constituye nuestra primera experiencia en la caracterización molecular de una familia cubana afectada con AME tipo I.
MÉTODOS
PRESENTACIÓN DEL CASO
Paciente masculino (II-2), de 2 meses de edad, nacido de embarazo a término, durante el cual la madre refiere movimientos fetales normales y parto a las 42 semanas, con circular apretada en el cuello. El llanto fue algo demorado y el peso de 380 g. Los padres tenían 22 años (ella) y 29 años (él) al nacimiento de éste, su segundo hijo, y no eran consaguíneos.Con pocos días de edad la madre le notó que era muy blandito, los movimientos de las extremidades eran muy pobres y el llanto débil. Como antecedentes de interés, la hermana mayor, producto del primer embarazo de la pareja, había fallecido a los 3 meses de edad con diagnóstico de enfermedad de Werdnig-Hoffmann, por lo que la pareja había asistido por asesoramiento con el genetista de su localidad, y conocidos los riesgos de recurrencia decidieron este segundo embarazo.
Examen físico: peso: 4,11 kg. Talla: 60 cm. Mirada alerta. Cráneo escafocefálico. Fontanela normotensa. Frente estrecha. Orejas de implantación baja con hiperenrollamiento del hélix bilate-ralmente. Desviación antimongoloidea de los ojos. Microrretrognatia. Fasciculaciones en la lengua. Anomalías de los pliegues de flexión palmar con clinodactilia del 5to. dedo en ambas manos. Surcos plantares profundos. Hipotonía muscular severa, disminución marcada de la fuerza muscular y arreflexia osteo-tendinosa generalizadas. Polipnea. Frecuencia respiratoria: 80/min. Tiraje intercostal bajo. Abundantes secreciones con dificultad respiratoria marcada.
Investigaciones realizadas: electromiograma: hallazgos compatibles con afectación del asta anterior de la médula espinal; biopsia muscular: atrofia de fibras musculares estriadas difusas. Se observan fibras aisladas hipertróficas. Atrofia muscular neurógena; electroencefalograma: normal; ultrasonido de cráneo: sistema ventricular normal.
Análisis del ácido desoxinucleótico (DNA): Se obtuvo DNA de la madre, el padre y el paciente de células nucleadas de sangre periférica en nuestro Laboratorio. En la Unidad de Genética Molecular referida, se amplificó por PCR y se genotiparon polimorfismos del tipo microsatélites. Cada individuo fue tipado para un número variable de polimorfismos suficientes como para conseguir informatividad total a ambos lados del locus SMA. Se tiparon preferentemente los marcadores más cercanos al locus SMA 5 y los más informativos. Se caracterizaron mutaciones en los genes para la proteína inhibidora de la apoptosis neuronal (gen NAIP) y de la supervivencia de la motoneurona (gen SMN).
RESULTADOS
Los resultados de las investigaciones electrofisiológicas e histopatológicas realizadas al paciente, confirmaron el diagnóstico de AME.En la figura aparecen representados los resultados del estudio genético-molecular realizado en la familia. En la región proximal al locus SMA se obtuvo informatividad completa para el microsatélite D5S1356 en los progenitores (ambos heterocigóticos). En el diplotipo materno fueron informativos los marcadores D5S557 y D5S610, mientras que en el paterno lo fueron D5S112 y D5S127. Se identificaron los haplotipos ligados con la enfermedad en el paciente y se encontraron deleciones de ambas copias del gen SMN (exón 8) y del gen NAIP (exón 5).
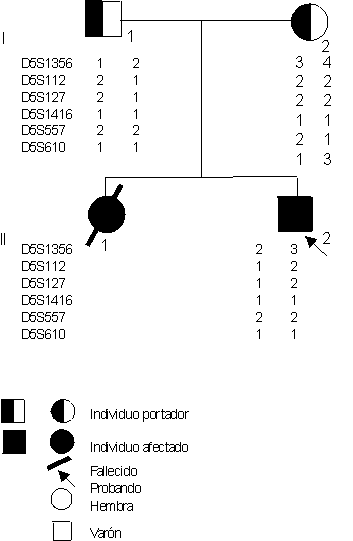
FIGURA. En ésta se muestran los haplotipos de los marcadores utilizados. En cada fila se indica el nombre del marcador y los 2 alelos de cada individuo. Cada columna representa un cromosoma. En el paciente (II 2) se observan los cromosomas paterno y materno asociados con la enfermedad.
DISCUSIÓN
El diagnóstico de enfermedad de Werdnig-Hoffmann tipo I se sospechó en el paciente por el inicio tan temprano de las manifestaciones clínicas y los antecedentes familiares. Se confirmó con los estudios electromiográficos e histopatológicos realizados.1,2,4 Como se conoce, es la forma más grave (aguda) de AME infantil. Los síntomas aparecen en los primeros meses de edad y fallecen durante el primer año de vida por fallo respiratorio.1,2 Afecta a 1 por cada 8 000 NV y tiene un riesgo de recurrencia de 25 %.6,9,10 Es causa frecuente de morbilidad y mortalidad infantil de algunos países.1-3El estudio molecular se realizó para identificar a los haplotipos ligados a la enfermedad y conocer el comportamiento del análisis mutacional directo de los genes SMN y NAIP. El primero se basó en estudios de ligamento y consistió en el análisis de los polimorfismos próximos al locus SMA. Este tipo de estudio proporciona un diagnóstico genético indirecto, pues mediante él se infiere indirectamente la presencia del gen afectado. Posee como requisito indispensable el diagnóstico preciso de la enfermedad de que se trate. En el paciente se identificaron los haplotipos o cromosomas parentales que se asocian con la enfermedad, y en este sentido la familia se puede considerar informativa como se muestra en la figura. Sin embargo, algunos marcadores resultaron homocigotos en el diplotipo materno o el paterno, lo que obviamente va a ocasionar dificultades en el manejo e interpretación del cromosoma afectado en caso de que exista entrecruzamiento en el resto de los marcadores. Esto podría resolverse con el tipado de otros microsatélites. Mientras mayor sea el número de marcadores que se investiguen, más confiables serán los resultados.3,5,8,12 De esta forma se va conformando la batería de marcadores más útiles para cada familia.
Hace poco se identificaron 2 genes en la región 5q13.1: el gen NAIP y el gen SMN. Los estudios realizados indican que ambos pudieran proporcionar el esperado diagnóstico genético directo de las AME.13-15 El gen de la proteína inhibidora de la apoptosis neuronal (NAIP) y el gen de la supervivencia de la motoneurona (SMN), obviamente también se localizan en 5q y se relacionan con las atrofias musculares espinales de la siguiente forma:12-15
- Han sido encontrados en sus 3 formas clínicas, y constituyen el fenómeno mutacional más frecuente las deleciones.
- Las deleciones del gen SMN no se correlacionan con la severidad o tipo clínico de la enfermedad, mientras que las deleciones del gen NAIP ocurren con más frecuencia en las AME tipo I.
- Las formas más ligeras de la enfermedad, muestran a su vez deleciones más pequeñas, las cuales incluyen predominantemente al gen SMN.
- La alta frecuencia de deleciones en el gen SMN sugiere que las mutaciones de él están directamente involucradas con la enfermedad, o que este gen yace muy cercano a la región crítica; al gen NAIP se le vincula con 2 posibles mecanismos: en la patogenia de las AME directamente, o interactuando con el producto génico del gen SMN.
Este trabajo es el primero de este tipo reportado en nuestro medio en un paciente con AME tipo I. Ilustra cómo el diagnóstico clínico debe preceder y es indispensable para la interpretación de los resultados y la correlación fenotipo-genotipo correspondiente. Hace énfasis en la importancia de realizar, oportunamente, el diagnóstico al nivel molecular en la enfermedad de Werdnig-Hoffmann, por ser el único que va a suministrar la información específica y necesaria para su detección prenatal.
SUMMARY
The infantile spinal muscular atrophies (SMA) are hereditary neuromuscular diseases characterized by degenerated Alfa-motoneurons of the anterior spinal marrow horn. Werdning-Hoffman disease, Type 1 SMA, is the most serious affection being transmitted as an autosomal recessive character, so those affected may die from respiratory failures in the first year of life. This paper presents a Cuban family with two kids who suffer from Type-1 SMA; the molecular analysis carried out in one of them identified two parental chromosomes responsible for the disease. Also, a deletion of both copies of SMN gene (exon 8) and NAIP gene (exon 5) were discovered in this patient. These findings showed the usefulness of this kind of studies with a view to making a prenatal diagnosis of Werdning-Hoffman disease.Subject headings: WERDNIG-HOFFMAN; EXONAS/genetics; GENETICS BIOCHEMICAL; CUBA.
REFERENCIAS BIBLIOGRÁFICAS
- Dubowitz V. Muscle disorders of childhood. London; Saunders;1978:146-190.
- Pearn JH. Classification of spinal muscular atrophies. Lancet 1980;i:919-22.
- Carter GT, Abresch RT, Fowler WM, Johnson ER, Kilmer D, McDonald C, et al. Profiles of neuromuscular diseases. Spinal Muscular Atrophies. Am J Phys Med Rehabil 1995;74(5):s150-9.
- Bono R, Inverno M, Botteon G, Lotti E, Etienne M, Berardinelli A, et al. Prospective study of gross motor development in children with SMA type II. Ital J Neurol Sci 1995;16(4):223-30.
- Munsat TL, Davies KE. Workshop Report. International SMA collaboration. Neuromusc Disord 1992;2:423-428.
- McKusick VA. Mendelian Inheritance in man. Catalogs of autosomal dominant, autosomal recessive, and X linked phenotypes. 11. ed. Baltimore: John Hopkins University; 1996.
- Brzustowicz LM, Lehner T, Castilla L, Penchaszadeh G, Wilkeman K, Daniels R, Davies K, et al. Genetic mapping of childhood-onset-spinal muscular atrophy to chromosome 5q11.2-11.3. Nature 1990;344:540-1.
- Gilliam TC, Brzustowicsa LM, Castilla LH, Lehener T, Penchaszadeh G, Daniels R, Byth B, et al. Genetic homogenety between acute and chronic forms of spinal muscular atrophy. Nature 1990;345:823-5.
- Melki J, Sheth P, Abdelahak S, Burlet P, Clermont O, Millaseau P, Reboullet S, et al. Mapping of acute (type I) spinal muscular atrophy to chromosome 5q12-q14. Lancet 1990;336:271-3.
- Wirth B, Pick E, Leutner A. Large linkage analysis in 100 families with autosomal recessive spinal muscular atrophy (SMA) and 11 CEPH families using 15 polimorphic loci in the region 5q11.2-q13.3. Genomics 1994;20:84-93.
- Mac Kenzie A, Roy N, Besner A, Mettler G, Jacob P, Korneluk R, Surh L, et al. Genetic linkage analysis of Canadian spinal muscular atrophiy kindreds using flanking microsatellite 5q13 polymorphisms. Hum Genet 1993;90:501-4.
- Hernández Chico C, Sampedro Velasco E, Valero Quirós C, Patiño García E, Moreno Herrero F. Diagnóstico prenatal de atrofia muscular espinal. Anales españoles de Pediatría 1995;42(6):429-35.
- Roy N, Mahadevan MS, McLean M, Shutler G, Yaraghi Z, Faraham R, Baird S, et al. The gene for neuronal apoptosis inhibitory protein is partially deleted in individuals with spinal muscular atrophy. Cell 1995;80:167-78.
- Lefebre S, Bürglen L, Reboullet S, Clermont D, Burlet P, Viollet L, Benichou B, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995;80:155-65.
- Rodríguez N, Owen N, Talbot K, Patel S, Muntoni F, Ignatius J, Dubowitz V, et al. Gene deletions in spinal muscular atrophy. J Med Genet 1996;33:93-6.
Dra. Ibis Menéndez Alejo. Hospital Pediátrico Docente "William Soler", San Francisco 10112, Altahabana, Ciudad de La Habana, Cuba.
1 Especialista de II Grado en Genética Clínica. Hospital Pediátrico Docente "William Soler".
2 Investigadora. Unidad de Genética Molecular. Hospital "Ramón y Cajal".
3 Especialista de I Grado en Neurología. Hospital Pediátrico Docente "William Soler".

