










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr v.70 n.2 Ciudad de la Habana abr.-jun. 1998
Xeroderma pigmentoso. Síndrome de Sanctis Cacchione. Presentación de 1 caso
Dra. Leopoldina Falcón Lincheta,1 Dra. Aida Dorticós Balea,2 Dr. Ramón Daniel Simón3 y Dr. Enrique Garbayo Otaño4Resumen
El xeroderma pigmentoso es una afección genética poco frecuente que traduce la hipersensibilidad celular a la radiación ultravioleta en asociación con una anormal reparación del ácido dexosirribonucleico, y que produce pecosidades, fotofobia y subsecuentemente cambios neoplásicos en zonas expuestas al sol. En este trabajo se presenta el primer caso reportado en Cuba en un niño de 5 años de edad, con la entidad clínica en su forma más severa de manifestarse, el síndrome de Sactis Cacchione, que involucra las manifestaciones cutáneas y oculares con trastornos neurológicos y somáticos graves, y provoca daños letales.Descriptores DeCS: XERODERMIA PIGMENTOSA; SINDROME DE COCKAYNE.
El xeroderma pigmentoso es una enfermedad cutánea de origen genético, con patrón de herencia autosómico recesivo, que se traduce por una hipersensibilidad marcada a las radiaciones ultravioletas, con aparición de lesiones semejantes a pecas, hiperpigmentación, queratosis, lesiones malignas y cicatrices atróficas limitadas en su inicio a las zonas expuestas a la luz solar, hasta posteriormente generalizarse; antes de los 30 años tienen un alto riesgo de incremento de carcinoma celular, de células escamosas o melanomas.1-5
Existen diferentes variedades de esta afección; la forma de presentación más severa se conoce como el síndrome de Sanctis-Cacchione, entidad que es muy poco frecuente en el mundo, pues sólo se han detectado menos de 60 casos. En Cuba no se había reportado ningún paciente con este síndrome.2-6
Sus características clínicas principales incluyen: deterioro progresivo del sistema nervioso con convulsiones, ataxia, espasticidad e incoordinación, hiporreflexia o arreflexia, microcefalia, trastorno del crecimiento moderado o importante, retraso mental, sordera neurosensorial, desarrollo sexual inmaduro, inteligencia baja y lesiones cutáneas en las regiones expuestas.1,3,4
Se postula que este síndrome se origina por un defecto en el proceso de escisión y reparación del ácido desoxirribonucleico (ADN) que presumiblemente es la base del deterioro de la piel tras la exposición a la luz ultravioleta. El pronóstico de esta forma clínica de xeroderma es grave, con peligro para la vida en etapas tempranas.
El diagnóstico prenatal es posible y se ha llevado a cabo en otros países.3,7,8
REPORTE DEL CASO
Se presenta un paciente que acude a consulta de nuestro centro hospitalario, de 5 años de edad, mestizo, natural de Santiago de Cuba; madre de 23 años, sana, con el único antecedente patológico familiar de una sobrina con sordomudez; padre de 27 años, sano, sin antecedentes patológicos familiares. Se detectó consanguinidad entre ambos (primos paternos). El paciente es el único hijo de este matrimonio nacido de un parto a término distócico, con un peso de 2 760 g, que presentó cianosis, por lo que se utilizaron maniobras de resucitación; llanto retardado, con buena succión y alimentación. La madre refiere que desde los 26 días de nacido comenzó a presentar una erupción eritematosa en regiones expuestas al sol, que luego se fueron tornando de color oscuro, y se generalizaron, con la aparición posterior de lesiones malignas tumorales.EXAMEN FÍSICO
Examen dermatológico. Presenta una erupción generalizada que respeta axilas y genitales externos, caracterizada por aumento de la pigmentación, entremezcladas con áreas de hipopigmentación y regiones con atrofia. Se observan también escamas y telangiectasias generalizadas. Hay múltiples cicatrices en antebrazos y cara, como secuelas de extirpación de carcinomas epidermoides (fig.).
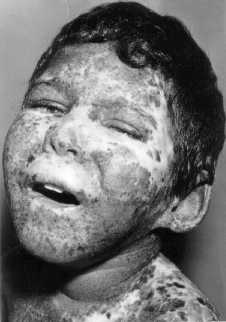
Examen oftalmológico. Presenta fotofobia, hendiduras parpebrales pequeñas en ambos ojos, hipertrofia de la conjuntiva dorsal a expensas de párpados inferiores, telangectasias conjuntivales bilaterales, mayor en ojo izquierdo, pigmentación redondeada de esclera cerca del limbo córneo-esclerar, córneas opacificadas en sus mitades inferiores y parcialmente vascularizadas, cámara anterior formada en ambos ojos, pupilas isocónicas con reflejos perezosos; al dilatar la pupila no se observaron sinequias; los medios acuosos, cristalino y vítreo, están transparentes.
Examen neurológico. Muestra retardo psicomotor, caminó a los 18 meses, primeras palabras a los 2 y medio años. Actualmente su lenguaje es muy escaso, explosivo y con dislalia. Presenta hiporreflexia generalizada y espasticidad que se acentúan en la marcha: sólo apoya la punta de los pies, además de ataxia, hiperquinesia marcada, microcefalia con circunferencia cefálica de 46 cm, respuesta plantar en abanico.
EXÁMENES COMPLEMENTARIOS QUE CORROBORARON SU DIAGNÓSTICO
Estudio psicológico. Por la escala de desarrollo de Gesell, se obtuvo un cociente de desarrollo general de 35, lo que implica un retardo severo.Electroencefalograma. Sueño inducido en etapa II, anormal, lento, bioccipital, inconstante con puntos lentos que se generalizan.
Estudio cromatográfico en orina. Se observa presencia de glicina.
Estudio cromatográfico en sangre. Se aprecia presencia de lisina en suero.
Estudio cromosómico. Normal.
Cultivo de tejidos. No se obtuvieron resultados por no haber crecimiento de la muestra.
Pruebas audiométricas. Sordera neurosensorial.
Examen histopatológico de la piel al microscopio de luz. Ligera hiperqueratosis, discreto edema focal intercelular de queratinocitos y células basales con incontinencia pigmentaria.
Examen histopatológico por microscopia electrónica. Piel hiperpigmentada; abundantes melanomas en estadio IV de maduración, alteraciones de forma y tamaño que están descritas en el xeroderma pigmentoso, piel hipopigmentada: melanocitos con escasez melanogénica y más pequeñas que la de la piel hiperpigmentada.
DISCUSIÓN
Los hallazgos verificados en el examen físico del paciente, así como la historia natural de la enfermedad y los resultados de los exámenes complementarios, apuntan claramente hacia el diagnóstico de este raro síndrome.La entidad ha sido reportada por todos los autores que la han descrito como de evolución tórpida y grave, en la que fallecen estos pacientes antes de la pubertad por neoformaciones.
El diagnóstico prenatal es posible y se ha llevado a cabo en otros países, al basarse en el hecho de que los fibroblastos normales de la piel, al cultivarlos, tienen la propiedad de reparar las escisiones, lo que está ausente en este trastorno, lo cual no se ha podido realizar en Cuba.
Para concluir diremos que:
- Los resultados del examen físico y de los complementarios, demuestran el diagnóstico del síndrome de Sanctis-Cacchione.
Teniendo en cuenta la no posibilidad actual del diagnóstico prenatal de esta afección en Cuba y su grave pronóstico, sólo un adecuado asesoramiento a los padres puede lograr que ellos adopten en lo adelante una correcta conducta reproductiva.
Summary
Xeroderma pigmentosum is a rare genetic affection that transforms cellular hypersensitivity to ultraviolet radiation in association with an abnormal repairing of the desoxyribonucleic acid, and that produces freckles, photophobia and, subsequently, neoplasitc changes in zones exposed to sunlight. In this paper it is reported the first case detected in Cuba in a five-year-old child suffering from the most severe manifestation of this disease, the Sanctis Cacchione´s syndrome, which involves skin and ocular manifestations with serious somatic and neurological disorders and provokes lethal damages.Subject headings: XERODERMA PIGMENTOSUM; COCKAYNE SYNDROME.
REFERENCIAS BIBLIOGRÁFICAS
- Kraemer KH. Heritable disease with increase sensitivity to cellular injury. En: Fitzpatrick TB. Eisen AZ, Wolff K, eds. Dermatology in general medicine. International Edition, 1993:1974-80.
- Goldsmith LA. Other disorder genetic skin. En: Emery A, Remoin David L. Principeis and practice of medical genetics. Edimburg: Livingstone, 1990:877-85.
- Lambert W. Genetic disease associated with DNA and chromosomal instability. Dermatol Clin 1987;5(1):85-95.
- Fernández Hernández-Baquero G. Genodermatosis. En: Dermatología. La Habana: Editorial Científico-Técnica, 1986:132-4.
- Day RS. Human adenoviruses as DNA repair probes. En: Nichols WW, Murphy DG, eds. DNA repair processes. Miami: Symposium Specialist, 1977:119-21.
- Mackie Rona M. Xerodermas pigmentosum. En: Emery Rimoin´s principles and practice of medical genetic. Edimburg: Livingstone, 1977:1325-6.
- Patterson JW, Jordan WP. Atypical fibroxanthoma in a patient with xeroderma pigmentosum. Arch Dermatol 1987;123(8):1066-70.
- Morison WL, Bucana C, Kripke ML. Impaired immune function in patients with xeroderma pigmentosum. Cancer Res 1985;45: 3929-31.
Dra. Leopoldina Falcón Lincheta. Avenida 41, número 2819, entre 28 y 34, reparto Kohly, municipio Playa, Ciudad de La Habana, Cuba.
1 Especialista de II Grado en Dermatología. Asistente en Dermatología.
2 Especialista de I Grado en Genética. Asistente en Genética.
3 Doctor en Ciencias. Asistente en Dermatología.
4 Especialista de I Grado en Dermatología.

