










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr v.77 n.1 Ciudad de la Habana ene.-mar. 2005
Presentaciones de casos
Hospital Pediátrico William Soler
Síndrome de Denys-Drash. Presentación de un caso
Dra. Neri Georgina Campañá Cobas,1 Dr. Sandalio Durán Alvarez,2 Dra. Yudamis Martínez Nieves,3 Dra. Nilvia Esther González García,4 Dra. Deborah A. García Martínez5 y Dr. Remigio Gómez Carrera 6
Resumen
El síndrome de Denys-Drash se caracteriza por pseudohemafroditismo masculino, tumor de Wilms y glomerulopatía con rápida progresión a la insuficiencia renal terminal, es producido por una mutación en el gen supresor TW1 localizado en el cromosoma 11p 13. La lesión glomerular se caracteriza por una esclerosis mesangial difusa. Reportamos un caso con genitales ambiguos, cariotipo 46 XY, síndrome nefrótico congénito a los 7 días de nacido, con rápida progresión a la insuficiencia renal terminal. Se hizo necesaria la diálisis peritoneal, y murió al mes de edad por sepsis generalizada. En el análisis del tejido renal se demuestra la esclerosis mesangial difusa.
Palabras clave: Síndrome de Denys-Drash, síndrome nefrótico congénito, insuficiencia renal en el recién nacido.
El síndrome de Denys-Drash (SDD) es una rara enfermedad genética que se caracteriza por pseudohemafroditismo masculino, tumor de Wilms y glomerulopatía.1,2 Puede presentarse como un síndrome nefrótico de inicio precoz con rápida progresión a la insuficiencia renal terminal y cuyas manifestaciones renales oscilan desde el período neonatal hasta la niñez. La herencia no está bien determinada ya que la mayoría de los casos adopta un carácter esporádico. Las mutaciones del gen TW ? parecen comportarse de manera dominante en lo que respecta a sus efectos sobre el sistema urogenital, pero de manera recesiva en el desarrollo del tumor de Wilms.3 Habib4,5 enfatizó en que los cambios histológicos del SDD están dados por una esclerosis mesangial difusa. La enfermedad sólo se ha reportado en nuestro país en un trabajo de enfermería,6 lo que nos motiva a publicar de este caso.
PRESENTACIÓN DEL CASO
Paciente nacido a las 37 semanas, peso 2 500 g, embarazo y parto normal, madre 37 años de edad, que cuando estaba ingresada en su localidad para estudio de genitales ambiguos, comienza a los 7 días de nacido con edema, oliguria, hipertensión arterial, proteinuria, hipoalbuminemia y elevación de los azoados. Se plantea el diagnóstico de un síndrome nefrótico congénito e insuficiencia renal crónica, y se remite para continuar su estudio en nuestro centro. Ingresó a los 13 días de edad, con fallo renal anúrico, edema generalizado (anasarca), y acidosis metabólica irreversible al tratamiento, por lo que es necesario implantar un cateter abdominal Tenkhoff y comenzar la diálisis peritoneal. Recibió además tratamiento con albúmina y antibiótico. Se plantea que el paciente es portador de un síndrome de Denys-Drash y se indicó su estudio. Al mes de edad comienza con dificultad para la diálisis, con elevación de los azoados, sepsis generalizada y fallece.
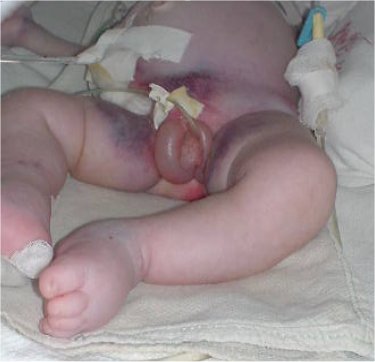
Al examen físico se evidencia edema generalizado y presencia de genitales ambiguos (figura 1).
Fig. 1. Genitales ambiguos. Estructura fálica que parece más larga y ancha que un clítoris pero más pequeña que un pene, con orificio superior, pliegues labioescrotales edematosos y fusionados en la parte superior
Complementarios:
Hemoglobina ---------------------------------------70 g / L
Creatinina -------------------------------------------280 µmol / L
Urea ------------------------------------------------17 µmol / L
Colesterol--------------------------------------------7 µmol / L
Triglicéridos-----------------------------------------2 µmol / L
Albúmina --------------------------------------------9,40 g / L
Proteínas totales-------------------------------------21,80 g / L
Cariotipo ---------------------------------------------46 XY
Cromatina sexual------------------------------------cuerpo de Barr negativo
Ultrasonido: Riñones aumentados de tamaño con hiperecogenicidad y mala delimitación córtico-medular.
La autopsia del paciente muestra la presencia de testículos pequeños, no descendidos, localizados en el abdomen por debajo del polo inferior de los riñones y microcalcificaciones en riñones, miocardio, cerebro, grasa perisuprarrenal y pulmones. En el análisis histológico del tejido renal se observa el desarrollo incompleto de algunos ovillos glomerulares, la esclerosis mesangial difusa del glomérulo con desarrollo completo y la dilatación quística de los túbulos renales con material proteínico intratubular (figura 2).
Fig. 2 Dilatación quística de los túbulos renales con material proteínico intratubular. Esclerosis mesangial difusa del glomérulo con desarrollo completo

DISCUSIÓN
Según la descripción original, el síndrome de Denys-Drash (SDD) está constituido por pseudohermafroditismo masculino, tumor de Wilms y glomerulopatía progresiva.2,7-9 Después de esa descripción original se encontraron pacientes con formas incompletas, y se dividió el síndrome en tres categorías: 1) genotipo masculino con las tres anormalidades, 2) genotipo masculino, nefropatía y genitales externos ambiguos, y 3) genotipo femenino con nefropatía y tumor de Wilms.3)
Clínicamente se caracteriza por proteinuria masiva con síndrome nefrótico en edad temprana de la vida y rápido progreso a la insuficiencia renal;4,10 la lesión hística es una esclerosis mesangial difusa (EMD).3,5 Desde el punto de vista clínico (genitales ambiguos, síndrome nefrótico con manifestaciones desde la segunda semana de la vida con rápida evolución a la insuficiencia renal) e histológico (EMD), nuestro paciente tiene las características de un síndrome de Denys-Drash (SDD). Los casos como el nuestro tienen alto riesgo de desarrollar posteriormente un tumor de Wilms.1
El defecto genital varía desde testículos no descendidos con hipospadias perineal hasta genitales externos femeninos normales con cariotipo 46XY, por lo que el estudio cromosómico debe realizarse en todos los pacientes con genitales externos femeninos que tengan un síndrome nefrótico con EMD.1 Nuestro paciente tenía cariotipo 46XY y en el estudio necrópsico se encontraron testículos intrabdominales normales .
En una revisión de 150 casos de SDD se encontró que 68 tenían genitales ambiguos. Según el fenotipo, 63 eran femeninos y 19 masculinos.11
En 1993 Habib reportó 36 pacientes con EMD. La nefropatía estaba aislada en 22 y en 14 la lesión estaba asociada bien a pseudohermafroditismo masculino o a tumor de Wilms.10 Cinco de estos pacientes presentaban los tres hallazgos del síndrome original y 9 sólo tenían dos componentes. De los 9 pacientes con tumor de Wilms, este era bilateral en 2, unilateral en 6 y en la autopsia de 1 paciente se encontraron nódulos nefroblásticos en un riñón. De 10 pacientes con pseudohermafroditismo masculino, 5 tenían asociado tumor de Wilms y todos tenían cariotipo 46XY. Ocho tenían genitales ambiguos con testículos disgenéticos, 1 tenía disgenesia gonadal mixta con un receptor de andrógeno parcialmente deficiente y 1 tenía un fenotipo femenino normal. En este último caso no se obtuvo información sobre las gónadas. Los 4 niños que sólo tenían tumor de Wilms eran femeninos según su fenotipo y en los 3 que se estudiaron se encontró cariotipo 46XY normal. Todos los pacientes, excepto uno, murieron o presentaron signos de insuficiencia renal crónica terminal antes de los 4 años de edad.10
Nuestro paciente, con genitales externos ambiguos, cariotipo 46XY normal y EMD, no tenía tumor renal detectable por ultrasonografía y en una búsqueda exhaustiva en el estudio necrópsico se descartó esta posibilidad. Sus testículos intrabdominales eran normales.
Ya que a que la enfermedad glomerular del SDD es resistente al tratamiento con diferentes drogas, el trasplante renal se mantiene como la única alternativa terapéutica que puede ser exitosa, y se han reportado resultados favorables sin recurrencia de la EMD.
Se sugiere la nefrectomía bilateral antes del trasplante renal por el riesgo de un tumor de Wilms oculto.2
Se denomina síndrome de Frasier (SF) a una condición similar desde el punto de vista clínico que se produce también por mutación en el gen del tumor de Wilms (WT1),12-15 sin embargo la lesión renal se caracteriza por glomeruloesclerosis focal y segmentaria (GFS).
Se ha sugerido que estos dos síndromes deben considerarse como parte del espectro de una misma enfermedad resultante de mutaciones del gen WT1 y no como dos enfermedades separadas. La clasificación clínica es importante para el pronóstico, ya que la enfermedad renal de base parece predecir la progresión de la glomerulopatía independientemente de la enfermedad genética. La evolución a la insuficiencia renal crónica terminal es más lenta cuando el paciente tiene una GFS como lesión de base.14
DENYS-DRASH'S SYNDROME. A CASE REPORT
Denys-Drash's syndrome is characterized by male pseudohermaphroditism, Wilms' tumor and glomerulopathy with fast progression to terminal renal failure. It is produced by a mutation in the TW1 suppressor gene located in the chromosome 11p 13. The glomerular lesion is characterized by a diffuse mesangial sclerosis. A case with ambiguous genitalia, 46 XY karyotype, and congenital nephrotic syndrome at 7 days of age, with fast progression to terminal renal failure, is reported. Peritoneal dialysis was necessary and the patient died at one month of age due to generalized sepsis. The diffuse mesangial sclerosis is showed in the analysis of the renal tissue.
Key words: Denys-Drash syndrome, congenital nephrotic syndrome, renal failure in the newborn infant.
Referencias bibliogrÁficas:
1. Barratt TM. Congenital nephrotic síndrome. En: Davison AM, Cameron JS, Grünfeld PJ, Kerr DNS, Eberhard R , Winearls CG, eds. Oxford Nephrology Clinic Oxford University Press; 1999.CD.ROM.
2. Holmberg C, Jalanko H, Tryguason K, Rapola J. Congenital nephrotic syndrome. En: Barratt TM, Avner ED, Harmon WE, eds. Pediatric Nephrology. 4 ed. Baltimore: Lippincott Williams and Wilkins; 1999: 765-777.
3. Rodríguez Soriano. Biología molecular de las glomerulopatías hereditarias. Panel de discusión. Disponible en: http://www.united.edu/cin2000/panelCin/conf/paneles50.html.
4. Habib R, Loirat C, Gubler MC, Niaudet P, Bensman A, Levy M, et al. The nephropathy associated with male pseudohermaphroditism and Wilms tumor (Drash syndrome): a distinctive glomerular lesion: report of 10 cases. Clin Nephrol 1985;24: 260-278.
5. Habib R, Gubler MC, Antigna CC, Loirat C, Gagnadoux MF. Síndrome néphrotique congénital ou infantile avec sclerose mesangiale diffuse. Ann Pediatr (Paris) 1990; 37: 73 – 77.
6. Izquierdo P, Figueroa M. Diagnóstico de enfermería identificados en un niño con el síndrome de Denys Drash. Rev Cubana de Enferm 2000; 16 (3): 185 – 8.
7. Denys P, Malvaux P, van den Berghe H, Tanghe W, Proesmans W. Association d'un syndrome anatomopathologique de pseuodohermafrodisme masculin, d'une tumeur de Wilms, d'une nephropathie parenchymatouse et d'un mosaicisme XX/XY, Arch Fr Pediatr 1967; 24: 729-739.
8. Drash A, Sherman F, Hartman W, Blizzard R. A syndrome of pseudohermaphroditism, Wilms tumor, hypertension, and degenerative renal disease. J Pediatr 1970; 76: 585-593.
9. Schumacher V, Schärer K, Wühl E, Altrogge H, Bonzel KE, Guschmann M, et al. Spectrum of early onset nephrotic syndrome associated with WTI missense mutations. Kidney Int 1998; 53: 1594-1800.
10. Habib R. Nephrotic syndrome in the 1st year of life. Pediatr Nephrol 1993; 7: 336. 346.
11. Mueller RF. The Denys-Drash syndrome. J Med Genet 1994; 31: 471-477.
12. Frasier SD, Bashore RA, Mosier HD. Gonadoblastoma associated with pure gonadal dysgenesis in monozygotic twins. J Pediatr 1964, 64: 740-745.
13. Moorthy AV, Chesney RW, Lubinsky M. Chronic renal failure and XY gonadal dysgenesis: "Frasier" syndrome a comentary on reported cases. Am J Med Genet 1987, 3: 297-301.
14. Demmer L, Primack W, Loik V, Brown R, Therville N, McRlreavey K. Frasier syndrome: A case of focal segmental glomerulosclerosis in a 46,XX female. J Am Soc Nephrol 1999; 10: 2215-2218.
15. McTaggart SA, Algar E, Chow CW, Powell HR, Jones CI. Clinical spectrum of Denys-Drash and Frasier syndrome. Pediatr Nephrol 2001; 16: 335-339.
Recibido: 3 de enero de 2005. Aprobado: 15 de febrero de 2005.
Dra. Neri Georgina Campañá Cobas. Servicios de Nefrología, Anatomía Patológica, Neonatología y Génetica del Hospital Pedíatrico William Soler. Calle 100 y Perla, Municipio Boyeros. Ciudad de La Habana.
1Especialista de I Grado en Pediatría. Diplomada en Nefrología.
2 Profesor consultante de Pediatría.
3Especialista de I Grado en Anatomía Patológica.
4Especialista de I Grado en Neonatología.
5Especialista de II Grado en Neonatología. J` Dpto. Genética.
6Residente de Pediatría.

