










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr v.77 n.2 Ciudad de la Habana jabr.-un. 2005
Hospital Pediátrico Universitario William Soler. Departamento de Neuropediatría
El síndrome de Landau-Kleffner. Presentación de dos casos
Dra. Albia J. Pozo Alonso,1 Dr. Desiderio Pozo Lauzán,2 Dra. Blanca Carrillo Valdés,3 Dra. Nitza Simón Chibás,4 Dra. Maritza Llanes Cepero5 y Dr. Desi Pozo Alonso6
Resumen
Se presentan dos niños que reúnen los criterios clínicos y electroencefalográficos del síndrome de Landau-Kleffner. En uno de los pacientes las dificultades para comprender el lenguaje y para la expresión oral espontánea comenzaron a manifestarse a los 6 años, y en el otro caso a los 6 años y 9 meses. Uno de los niños no ha presentado crisis epilépticas hasta el momento actual. En este paciente la tomografía computadorizada por emisión de positrón único (SPECT cerebral) mostró una ligera hipoperfusión temporoparietal posterior bilateral. En el niño que presenta crisis epilépticas; éstas se iniciaron a la edad de 2 años y 6 meses y eran focales simples motoras que se generalizaron secundariamente. Desde hace un año no presenta crisis. Desde el punto de vista clínico y electroencefalográfico, ambos pacientes tuvieron una respuesta favorable al tratamiento con prednisona. En un niño, aunque ha mejorado, persisten las dificultades para la comprensión y la expresión orales. Se concluye que son muy importantes para el diagnóstico de este síndrome la existencia de una afasia adquirida y las descargas observadas en el electroencefalograma.
Palabras clave: Afasia epiléptica adquirida, agnosia auditiva verbal, encefalopatía epiléptica, síndrome de Landau-Kleffner.
El síndrome de Landau-Kleffner es de rara aparición y se presenta en el 0,2 % de las epilepsias en la infancia.1 De acuerdo con el nuevo esquema de diagnóstico propuesto para personas con crisis epilépticas y epilepsias,2 se ubica entre las encefalopatías epilépticas en las que, se piensa, las anomalías epileptiformes contribuyen al trastorno progresivo de la función cerebral.
Este síndrome se caracteriza por la asociación de afasia adquirida, por lo general de tipo receptivo, y un trazado electroencefalográfico en el que se observan descargas de puntas y puntas-ondas focales, multifocales y generalizadas, que se incrementan durante el sueño.3 Predomina en el sexo masculino4 y fue descrito por Landau y Kleffner en 1957.5
Las crisis epilépticas y los trastornos de conducta son frecuentes, aunque no están presentes en todos los niños.3,4,6 En estos pacientes se ha observado hipercinesia, estallidos de ira, rebeldía, manifestaciones agresivas y rasgos autistas.3 En algunos casos se manifiesta disfunción motora con pérdida de la destreza manual.7
Se desconocen las causas de este síndrome, sin embargo se ha planteado que el mecanismo autoinmunitario y algunas infecciones, como las producidas por el virus del herpes simple, pudieran estar involucradas en su origen.7
Nos propusimos con este trabajo presentar 2 pacientes del sexo masculino que reúnen los criterios clínicos y electroencefalográficos de esta encefalopatía epiléptica.
CASOS CLÍNICOS
Caso 1
Paciente del sexo masculino, cuya familia refiere que a la edad de 6 años y 9 meses no responde a las preguntas, como si no escuchara lo que le dicen. El paciente manifiesta disminución de la expresión oral espontánea y un lenguaje ininteligible. En la escuela presenta dificultades para escribir al dictado, pero no para los cálculos matemáticos.
Antecedentes prenatales y perinatales:
- Embarazo normal. Parto distócico por cesárea a las 38 semanas. Apgar 9/9.
- Neurodesarrollo normal.
- Antecedentes patológicos personales: dislalia.
- Antecedentes patológicos familiares: en el padre, asma bronquial.
- Examen físico general, regional y por sistemas normal.
- Examen físico neurológico: en ocasiones no comprende lo que se le dice. Con frecuencia no responde a nuestras preguntas. Lenguaje ininteligible. Hipercinesia.
Exámenes complementarios:
- Potenciales evocados auditivos de tallo cerebral normales.
- Electroencefalograma (EEG) de vigilia lento de base. Descargas de puntas y puntas-ondas occipitales izquierdas, también en región central derecha, parietales y occipitales derechas , en región temporal media derecha y en vertex anterior (fig 1).
- EEG con privación de sueño de 24 horas con registro en las etapas I, II y III de sueño. En esta última etapa las descargas se hicieron más frecuentes y continuas con respecto al trazado de vigilia (fig 2).
- Tomografía computadorizada por emisión de positrón único (SPECT): ligera hipoperfusión temporo-parietal posterior bilateral.
- Tomografía axial computadorizada de cráneo (TAC) normal.

Figura 1. Caso 1. EEG de vigilia multifocal

Figura 2. Caso 1. EEG de sueño en etapa III. Las descargas de puntas-ondas
se hicieron más frecuentes y continuas con relación al trazado de vigilia
Se inicia el tratamiento con prednisona oral en dosis de 2 mg/kg/día. Al mes de comenzado el tratamiento se realiza un EEG con privación de sueño de 24 horas en etapa III, en el que desaparecieron las descargas (fig 3). Clínicamente se constata mejoría con relación a la comprensión del lenguaje y la expresión oral, aunque la recuperación no es total.
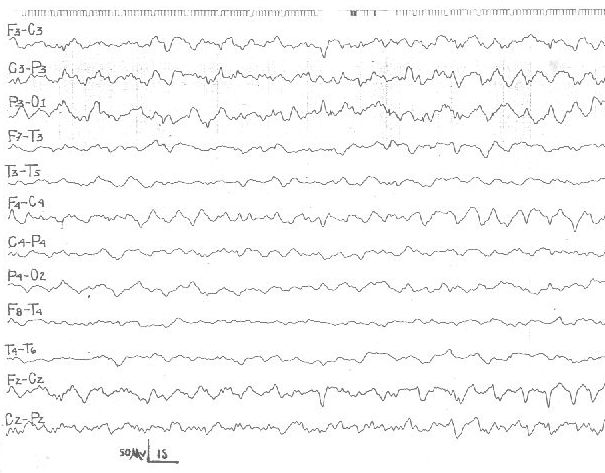
Figura 3. Caso 1. EEG de sueño en etapa III. Se observa la desaparición
de las descargas después de un mes de iniciado el tratamiento con prednisona
Caso 2
Paciente del sexo masculino que comenzó a presentar, a la edad de 6 años, un lenguaje espontáneo escaso. Los familiares tienen la impresión que el niño no comprende lo que le dicen, no responde a las preguntas ni sostiene una conversación. A los 2 años y 6 meses comenzó a presentar crisis epilépticas focales simples motoras izquierdas que se generalizan secundariamente. Se le inició tratamiento con fenitoína y desde hace un año no tiene crisis.
Antecedentes prenatales y perinatales:
- Embarazo y parto normales. Apgar 9/9.
- Neurodesarrollo normal.
- Antecedentes patológicos personales: epilepsia focal desde los 2 años y 6 meses.
- Antecedentes patológicos familiares: un hermano, un tío y primo presentan epilepsia.
- Examen físico neurológico: no presenta lenguaje espontáneo, no mantiene una conversación, no comprende lo que se le dice.
Exámenes complementarios:
- EEG de vigilia multifocal.
- EEG con privación de sueño de 24 horas: se obtiene trazado de sueño multifocal en etapa III.
- Potenciales evocados auditivos de tallo cerebral normales.
- TAC de cráneo normal.
- Resonancia magnética nuclear cerebral (RMN): moderados signos de hemiatrofia del hemisferio cerebral izquierdo con predominio temporal, frontal y basal.
Se inicia tratamiento con prednisona oral a razón de 2 mg/kg/día. A las 2 semanas de tratamiento los familiares constatan mejoría de la comprensión y del lenguaje espontáneo. A los 2 meses de iniciado el tratamiento con prednisona el niño comprende todo lo que se le dice y ha reaparecido el lenguaje espontáneo.
El electroencefalograma con privación de sueño de 24 horas evolutivo se registra en etapa III de sueño. La frecuencia de las descargas es menor, fundamentalmente en el hemisferio izquierdo. Continúa observándose un foco muy activo de descargas de puntas rápidas en temporal medio derecho. Es notable la disminución de la amplitud en comparación con el EEG anterior.
DISCUSIÓN
Las manifestaciones clínicas del síndrome de Landau-Kleffner se inician entre los 18 meses y los 13 años3 aunque la máxima incidencia se encuentra entre los 5 y 7 años.8 La afasia está presente en todos los pacientes y constituye la primera manifestación en el 50 % de los casos. Ocurre en niños previamente normales aunque en algunos casos el desarrollo del lenguaje puede haber sido algo retrasado.3 En la serie de Caraballo y otros,9 la edad media de comienzo de la afasia fue de 4,8 años y la edad media de inicio de las crisis epilépticas, de 5 años. En todos los pacientes que presentaron crisis epilépticas, éstas comenzaron antes o simultáneamente con el inicio de la afasia.
En uno de nuestros pacientes las crisis epilépticas se iniciaron a los 2 años y 6 meses y posteriormente a los 6 años desarrolló la afasia. En el otro las dificultades para la comprensión del lenguaje y para la expresión oral espontánea comenzaron a los 6 años y 9 meses.
El inicio de la afasia es generalmente subagudo6 o progresivo, y se instaura en el transcurso de días o semanas.3 El tipo de afasia que se observa es una agnosia verbal auditiva. El paciente comienza a presentar dificultades para comprender el lenguaje, más tarde tiene dificultades en la articulación y disminuye la expresión oral espontánea.6 El mutismo es frecuente.3
Las crisis epilépticas se presentan en el 72 % de los casos y, aunque pueden manifestarse antes o después del inicio de la afasia,9 en el 20 % de los pacientes la primera de ellas ocurre durante la instauración progresiva de la agnosia auditiva.4 Las crisis epilépticas repetitivas son raras, ocurren fundamentalmente en horario nocturno y por lo general se controlan bien con los medicamentos antiepilépticos.4
Los tipos de crisis epilépticas más frecuentes son las clónicas generalizadas, focales simples motoras, ausencias atípicas y crisis unilaterales; 6 las crisis parciales complejas son infrecuentes.4 En el estudio de Caraballo y otros,9 las crisis epilépticas más frecuentes fueron las mioclonias palpebrales, ausencias atípicas y las crisis focales simples motoras. Uno de nuestros pacientes sólo ha presentado hasta el momento actual crisis focales simples motoras que se generalizan secundariamente.
Se han observado antecedentes familiares de epilepsia en el 12 % de los casos.4 En los 4 pacientes estudiados por Takeoka y otros,10 no se encontraron antecedentes familiares de epilepsia. Uno de nuestros pacientes tiene varios familiares con crisis epilépticas.
En este síndrome, el EEG de vigilia muestra puntas y puntas-ondas de gran amplitud, repetitivas, las cuales se localizan en las regiones temporales y parieto-occipitales.4 También se han observado descargas de puntas-ondas generalizadas.11 El sueño, fundamentalmente al inicio, aumenta las anomalías epilépticas. En ocasiones, durante la evolución del síndrome de Landau-Kleffner, el EEG de sueño muestra un patrón de puntas-ondas continuas bilaterales que ocupan más del 85 % del sueño lento, lo que representa un estado de mal electroencefalográfico durante el sueño lento. Tras este hallazgo electroencefalográfico, se ha planteado la hipótesis de que el síndrome de Landau-Kleffner constituye una variante clínica de la encefalopatía con estado de mal eléctrico durante el sueño.12
En uno de nuestros pacientes, el primer EEG de sueño, realizado después de privación de sueño de 24 horas, mostró un incremento de la frecuencia de las descargas con relación al EEG de vigilia.
En estos pacientes las pruebas audiométricas son normales,13 al igual que los potenciales evocados auditivos de tallo cerebral.14 Sin embargo, pueden encontrarse anormalidades en los potenciales evocados auditivos corticales, lo que sugiere que en este síndrome se afectan los mecanismos que intervienen en el procesamiento de la información auditiva verbal.15 En nuestros dos pacientes fueron normales los potenciales evocados auditivos de tallo cerebral
La patogénesis del síndrome de Landau-Kleffner no se ha definido. Se ha planteado que podría deberse a una alteración del encéfalo responsable de la epilepsia y de la afasia, o a una alteración funcional inducida directamente por la interferencia sobre el lenguaje de la actividad epiléptica.3
La mayoría de los estudios neuroimagenológicos (TAC y RMN) resultan normales,9 aunque se han encontrado en algunos pacientes lesiones focales como cisticercosis,16 vasculitis,17 tumores18 y enfermedad desmielinizante inflamatoria.19 En la comunicación de Pablo y otros7 se observó en la TAC de cráneo un quiste en la línea media. En uno de nuestros pacientes, la RMN de cráneo mostró moderados signos de hemiatrofia cerebral izquierda con predominio temporal, frontal y basal.
En 4 niños con síndrome de Landau-Kleffner se realizaron análisis volumétricos mediante resonancia magnética de varias regiones neocorticales y estructuras subcorticales en los que se detectó una disminución del volumen cortical en las áreas temporales superiores bilaterales, que corresponde con la corteza de asociación auditiva. En 2 niños la disminución del volumen cortical fue mayor en la zona con más actividad epileptiforme.10 Recientemente, Bourgeois y Landau8 comentan estos hallazgos y sugieren que la atrofia cortical encontrada pudiera ser el resultado de la actividad epileptiforme refractaria y explicaría por qué la recuperación completa del lenguaje a largo plazo es con frecuencia pobre, a pesar del control de las crisis y la recuperación en el EEG. Sugieren la hipótesis de que la actividad epileptiforme del lóbulo temporal provoca las crisis epilépticas y los trastornos del lenguaje. La excitoxicidad de la actividad epiléptica provoca atrofia cortical focal y ésta a su vez impide la recuperación del lenguaje a pesar de la desaparición de la actividad epileptiforme.
Estudios mediante tomografías por emisión de positrón (PET)20 y tomografías computadorizadas por emisión de positrón único (SPECT)21,22 han evidenciado anormalidades en las áreas temporales. En uno de nuestros pacientes la SPECT mostró ligera hipoperfusión temporo-parietal posterior bilateral.
Se han empleado diferentes medicamentos para el tratamiento del síndrome de Landau-Kleffner. Los esteroides y la hormona adrenocorticotropa (ACTH) han resultado eficaces pues mejoran de forma importante y más prolongada, todas las manifestaciones del síndrome.4 Se han comunicado resultados favorables con el empleo de inmunoglobulinas por vía endovenosa.23,24 El ácido valproico, la etosuximida y las benzodiazepinas han resultado beneficiosos pero sus efectos son parciales y transitorios en muchos casos.4,25 La fenitoína, la carbamazepina y el fenobarbital son ineficades e incluso pueden agravar el cuadro clínico y electroencefalográfico.25 La vigabatrina puede aumentar las anomalías en el EEG.3 El tratamiento quirúrgico mediante la técnica de transección subpial múltiple se ha realizado en algunos casos,26 en los que se ha observado mejoría de las funciones del lenguaje.
La estimulación del nervio vago es otro recurso terapéutico. Se empleó en 6 pacientes con el síndrome y a los 6 meses de haberla realizado, 3 de los casos experimentaron una disminución en la frecuencia de las crisis.27 El tratamiento de rehabilitación del lenguaje se debe realizar siempre.3
Nuestros 2 pacientes recibieron tratamiento con prednisona oral a razón de 2 mg/kg/día. Al mes de iniciado el tratamiento en uno de los pacientes se constató mejoría clínica en la comprensión del lenguaje, la expresión oral y desaparecieron las descargas en el EEG. En el otro paciente, a los dos meses de iniciado el tratamiento con prednisona, reapareció el lenguaje espontáneo y fue capaz de mantener una conversación. A pesar de que en el EEG persistían las descargas, se observó una disminución de la frecuencia de estas. Ambos pacientes recibieron tratamiento de rehabilitación del lenguaje con seguimiento frecuente por el Departamento de Logopedia y Foniatría de nuestro hospital.
La afasia se caracteriza por un curso fluctuante, con etapas de remisión y de agravación.11 En un estudio evolutivo realizado con un mínimo de seguimiento de 7 años,28 se pudo constatar que el 47,5 % de los casos carecían de expresión oral o presentaban un lenguaje oral ininteligible. Se ha planteado que el pronóstico es mejor, cuando la pérdida del lenguaje ocurre después de la edad de 6 años y cuando los tratamientos medicamentoso y logopédico se inician precozmente.11 En muchos casos la recuperación total es inusual y persisten dificultades variables en la comunicación verbal.3 El pronóstico de la epilepsia es favorable.4
AGRADECIMIENTOS
Agradecemos al Departamento de Imagenología del Hospital CIMEQ y al Departamento de SPECT del Centro Internacional de Restauración Neurológica (CIREN) la colaboración brindada para la realización de este trabajo.
Summary
Landau-Kleffner's syndrome
Two children with the clinical and electroencephalographic criteria of Landau-Kleffner's syndrome are presented. In one of the patients, the difficulties for understanding the language and for the spontaneous oral expression started at 6 years old. In the other case, they were manifested at 6 years and 9 months old. One of the children has not had epileptic seizures so far. In this patient, the single positron emission computerized tomography (brain SPECT) showed a mild bilateral posterior temporoparietal hypoperfusion. The epileptic seizures in the other child started when he was 2 years and 6 months old and were focal , simple, motor and secondarily generalized. He has not had seizures for a year. Both patients had a favorable response to prednisone from the clinical and electroencephalographic point of view. The difficulties for understanding and oral expression persist in a child, although he has improved. It is concluded that the existence of an acquired aphasia and the discharges observed in the electroencephalogram are very important for the diagnosis of this syndrome.
Key words: Acquired epileptic aphasia, verbal auditive agnosia, epileptic encephalopathy, Landau-Kleffner's syndrome.
REFERENCIAS BIBLIOGRÁFICAS
1. Kramer U, Nevo Y, Neufeld MY, Fatal A, Leitner Y, Harel S. Epidemiology of epilepsy in childhood: a cohort of 440 consecutive patients. Pediatr Neurol 1998; 18:46 -50.
2. Engel J Jr. A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: report of the ILAE Task Force on Classification and Terminology. Epilepsia 2001;42:1-8.
3. Aicardi J. El síndrome de Landau-Kleffner. Rev Neurol 1999;29:380-5.
4. Beaumanoir A. Le syndrome de Landau-Kleffner. En: Roger J, Bureau M, Dravet Ch, Dreifuss FE, Perret A, Wolf P, eds. Les syndromes épileptiques de l'enfant et de l'adolescent. 2 ème edn.London: John Libbey; 1992. p.231-44.
5. Landau WM, Kleffner FR. Syndrome of acquired aphasia with convulsive disorder in children. Neurology 1957;7:523-30.
6. Tassi nari CA, Rubboli G, Volpi L, Billard C, Bureau M. Etat de mal électrique épileptique pendant le sommeil lent (ESES ou POCS) incluant l'aphasie épileptique acquise (syndrome de Landau-Kleffner). En: Roger J, Bureau M, Dravet Ch, Genton P, Tassinari CA, Wolf P, eds. Les syndromes épileptiques de l'enfant et de l'adolescent. 3ème éd. London: John Libbey, 2002. p.265-83.
7. Pablo MJ, Valdizán JR, Carvajal P, Bernal M, Peralta P, Sáenz de Cabezón A. Síndrome de Landau-Kleffner. Rev Neurol 2002;34:262-4.
8. Bourgeois BFD, Landau WM. Landau-Kleffner syndrome and temporal cortical volume reduction. Cause or effect? Neurology 2004;63:1152-3.
9. Caraballo RH, Yépez II, Soprano AL, Cersósimo RO, Medina C, Fejerman N. Afasia epiléptica adquirida. Rev Neurol 1999;29:899-907.
10. Takeoka M, Riviello JJ, Duffy FH, Kim F, Kennedy DN, Makris N, et al. Bilateral volume reduction of the superior temporal areas in Landau-Kleffner syndrome. Neurology 2004;63:1289-92.
11. Hirsch E, Marescaux C, Maquet P, Metz-Lutz MN, Kiesmann M, Salmon E, et al. Landau-Kleffner syndrome: a clinical and EEG study of five cases. Epilepsia 1990;31:756-67.
12. Tassinari CA, Michelucci R, Forti A, Salvi F, Plasmati R, Rubboli G, et al. The electrical status epilepticus syndrome. En: Degen R, Dreifuss FE ,eds. Benign localized and generalized epilepsies of early childhood. Amsterdam :Elsevier, 1992(a). p.11-5.
13. Rapin I, Mattis S, Rowan AJ, Golden GG. Verbal auditory agnosia in children. Dev Med child Neurol 1977;19:192-7.
14. Nakano S, Okuno T, Mikawa H. Landau-Kleffner syndrome: EEG topographic studies. Brain Dev 1989;11:43-50.
15. Fejerman N, Medina C, Caraballo R. Trastornos paroxísticos y síntomas episódicos:epilepsias. En: Fejerman N, Fernández-Alvarez A, eds. Neurología Pediátrica. 2ed. Buenos Aires: Panamericana, 1997. p.531-73.
16. Otero E, Córdova S, Díaz F, García-Terrel I, del Bruto O. Acquired epileptic aphasia (Landau-Kleffner syndrome) due to neurocysticercosis. Epilepsia 1989;30:569-72.
17. Pascual-Castroviejo I, López-Martín L, Martínez-Bermejo A , Pérez-Higueras A. Is cerebral arteritis the cause of the Landau-Kleffner syndrome? Four cases in childhood with angiographic study. Can J Neurol Sci 1992; 19:46 -52.
18. Solomon G, Carson D, Pavlakis S, Fraser R, Labar D. Intracranial EEG monitoring in Landau-Kleffner syndrome associated with a temporal lobe astrocytoma. Epilepsia 1993; 34:557-60.
19. Perniola T, Magari L, Buttiglione M, Andreula C, Simone IL, Santostasi R. A case of Landau-Kleffner syndrome secondary to inflammatory demyelinating disease. Epilepsia 1993;39:551-6.
20. Da Silva EA, Chugani DC, Muzik O, Chugani HT. Landau-Kleffner syndrome: metabolic abnormalities in temporal lobe are a common feature. J Child Neurol 1997;12:489-95.
21. Harbord MG, Singh R, Morony S. SPECT abnormalities in Landau-Kleffner syndrome. J Clin Neurosci 1999;6:9-16.
22. Raybarman C. Landau-Kleffner syndrome: a case report. Neurol India 2002;50:212-3.
23. Lagae LG, Silberstein J, Gilhs PL , Casaer PJ. Successful use of intravenous immunoglobulin in Landau-Kleffner syndrome. Pediatr Neurol 1998;18:165-8.
24. Mikati MA, Saab R. Successful use of intravenous immunoglobulin as initial monotherapy in Landau-Kleffner syndrome. Epilepsia 2000;41:880-6.
25. Marescaux C, Hirsh E, Finck S, Marquet P, Shlumberger E, Sellae F, et al. Landau-Kleffner syndrome. A pharmacologic study of five cases. Epilepsia 1990;31:768-77.
26. Morrell F, Whisler WW, Smith MC, Hoeppner TJ, De Toledo-Morrell L, Pierre-Louis SJ, et al. Landau-Kleffner syndrome. Treatment with subpial intracortical transection. Brain 1995;118:1529-46.
27. Park YD. The effects of vagus nerve stimulation therapy on patients with intractable seizures and either Landau-Kleffner syndrome or autism. Epilepsy Behav 2003;4:286-90.
28. Dugas M, Gérard CL, Franc S, Lecendreux M. Late onset acquired epileptic aphasia. En: Beaumanoir A, Bureau M, Deonna T, Mira M, Tassinari CA, eds. Continous spikes and waves during slow sleep. Electrical status epilepticus during slow sleep. London: John Libbey, 1995. p.143-7.
Recibido: 9 de mayo de 2005. Aprobado: 15 de julio de 2005.
Dra Albia J. Pozo Alonso. Calle 100 y Perla. Altahabana, Boyeros. Ciudad de La Habana. Cuba.
Correo electrónico: albiap@infomed.sld.cu
1Profesora Auxiliar. Especialista de II Grado en Pediatría y Neurología.
2Especialista de II Grado en Neurología y Pediatría. Doctor en Ciencias Médicas. Profesor Titular.
3Especialista de I Grado en Otorrinolaringología.
4Especialista de I Grado en Medicina General Integral y en Logopedia y Foniatría.
5Especialista de I Grado en Psiquiatría.
6Especialista de I Grado en Medicina General Integral.

