










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr v.78 n.2 Ciudad de la Habana abr.-jun. 2006
Hospital Pediátrico Universitario «William Soler»
Hiperostosis cortical infantil. A propósito de tres casos
Dra. Neri G. Campañá Cobas,1 Dr. Julio Santana Garay,2 Dr. Sandalio Duran Álvarez,3 Dra. Belkis Vázquez Ríos4 y Dr. Horacio Moreno Díaz5
RESUMEN
La hiperostosis cortical infantil o síndrome de Caffey-Silverman es un trastorno de causa desconocida que afecta al esqueleto y a algunas de las fascias contiguas y músculos. Presentamos tres pacientes que ingresaron en nuestro hospital por presentar irritabilidad, fiebre y tumefacción dolorosa en ambos lados de la mandíbula (dos pacientes) o en el lado izquierdo (un paciente), eritrosedimentación acelerada, aumento de la fosfatasa alcalina y anemia. Se concluye que los tres pacientes presentan un síndrome de Caffey-Silverman.
Palabras clave: Hiperostosis, síndrome de Caffey, irritabilidad, fiebre.
La hiperostosis cortical infantil (HCI) o síndrome de Caffey-Silverman es una enfermedad autolimitada, de etiología desconocida, que se presenta en los primeros meses de la vida e incluso se han reportado casos intraútero.1-3 Se caracteriza por un trastorno que afecta al esqueleto y a algunas de sus fascias contiguas y músculos. Se presenta en todas las circunstancias, en ciudades, comunidades rurales, en todo tipo de clima, en todas las estaciones, en todo tipo racial, en todos los estratos sociales y con igual incidencia para uno y otro sexo.1,2
La presentación clásica de la enfermedad es irritabilidad, tumefacción de los tejidos blandos y engrosamiento cortical del hueso subyacente. La HCI ha sido demostrada en todos los huesos tubulares del esqueleto, pero los que se afectan con mayor frecuencia son la mandíbula y las clavículas.1,2,4 La presentación de la forma prenatal se caracteriza por polihidramnios, hipoplasia pulmonar, hepatomegalia, prematuridad y alta letalidad.3, 5
El diagnóstico se establece sobre la base de la historia clínica y los hallazgos radiográficos. No existe ningún hallazgo patognomónico de la enfermedad. Tiene un curso benigno, las manifestaciones remiten sin tratamiento en varias semanas o meses. No hay tratamiento específico.
Por lo raro de la enfermedad hemos querido presentar estos tres pacientes, para recordar su diagnóstico y conducta.
PRESENTACIÓN DE LOS CASOS
|
| Caso 1 | Caso 2 | Caso 3 |
| Edad | 2 meses | 3 meses | 2 meses |
| Raza | Blanca | Negra | Mestiza |
| Sexo | Masculino | Masculino | Femenino |
| APP | Lactancia mixta, EDA | Prematuro, EDA, | Lactancia materna, |
| MI | Irritabilidad y fiebre | Irritabilidad y fiebre | Irritabilidad y fiebre |
| Examen | Palidez cutáneo-mucosa, hipotonía hiperreflexia, a los 15 días tumoración en ambos lados de la | Palidez cutáneo-mucosa, hipotonía hiporeflexia osteo-tendinosa, a los 13 días tumoración de la mejilla izquierda. | Palidez cutáneo-mucosa, hipotonía, a los 12 días aumento de volumen difuso de ambos lados de la cara.(fig.) |
APP: antecedetes patológicos personales; MI: motivo de ingreso; EDA: Enfermedad diarreica aguda.
El cuadro siguiente ilustra los exámenes de laboratorio realizados a cada uno de los casos:
|
| Caso 1 | Caso 2 | Caso 3 |
| Hb | 71 g/L | 74 g/L | 88 g/L |
| Eritro | 140 mm/h | 78 mm/h | 125 mm/h |
| Leucocitos | Leucocitosis ligera | Normal | Leucocitosis moderada |
| Lámina periférica | Anisocitosis, poiquilocitosis, | Anisocitosis, gránulos toxicos | Anisocitosis, poiquilocitosis, gránulos tóxicos |
| LCR | Negativo | Negativo | Negativo |
| Fosfatasa A | 349,5 UI | 400 UI | 734 UI |
| Hemocultivo | Negativo | Negativo | Negativo |
| Cituria | Negativa | Negativa | Negativa |
| Urocultivo | Negativo | Negativo | Negativo |
Hb: hemoglobina; LCR: líquido cefalorraquídeo; A: alcalina.
Los ultrasonidos y las radiografías arrojaron los resultados siguientes:
- Caso 1. El ultrasonido de la cara mostró aumento de volumen de partes blandas en ambos lados del maxilar. La radiografía de tórax fue negativa. La vista anterior de la radiografía del maxilar inferior evidenció un aumento de volumen de las partes blandas y engrosamiento ligero del periostio.
- Caso 2. El ultrasonido de la cara mostró aumento de volumen de partes blandas, de aspecto difuso en el lado izquierdo del maxilar inferior. La radiografía de tórax fue negativa. La vista anterior de la radiografía del maxilar inferior evidenció un aumento de volumen de las partes blandas del lado izquierdo.
- Caso 3. El ultrasonido de la cara mostró aumento de partes blandas de aspecto difuso de ambos lados del maxilar. La radiografía de tórax fue negativa. La vista anterior de la radiografía del maxilar inferior evidenció un aumento de volumen de las partes blandas y engrosamiento del periostio (figura)
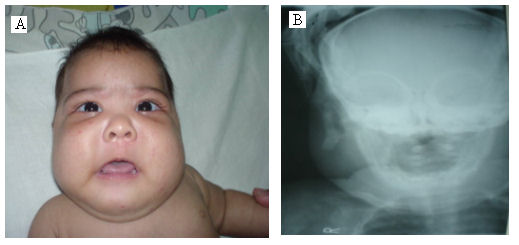
Figura.
El caso 1 recibió un tratamiento con cefalosporina de tercera generación al inicio (7 días), seguido de aspirina durante un mes. La evolución fue favorable. Al mes se normalizó la eritrosedimentación y la hemoglobina aumentó. La tumoración desapareció a los 2 meses y las lesiones óseas después del año.
El caso 2 recibió una cefalosporina de tercera generación al inicio (7 días). La evolución fue favorable. La eritrosedimentación y la hemoglobina se normalizaron a los 22 días y la tumoración desaparece al mes.
El caso 3 también recibió una cefalosporina de tercera generación durante 7 días y esteroides durante 15 días. La evolución fue favorable. La eritrosedimentación y la hemoglobina se normalizaron a los 4 meses. Las lesiones óseas no han desaparecido después de 9 meses y tuvo una recaída a los 10 meses.
DISCUSIÓN
La hiperostosis cortical infantil (HCI) fue descrita en 1945 en forma independiente por Caffey, Silverman, Smith y colaboradores como un engrosamiento cortical poco común de ciertos huesos de los lactantes.1,2 Aunque ya había sido reconocida en 1930 por Roske y por Ellis en 1938.6
Es una enfermedad autolimitada que se presenta en los primeros meses de la vida, por lo general antes de los 6 meses de edad, de etiología desconocida, aunque se describe la existencia de dos formas: la esporádica y la familiar.5
Se han sugerido varias teorías: a) que puede ser una osteodisgenesia embrionaria consecuencia de un defecto local del riesgo sanguíneo del área; b) que exista un defecto hereditario de las arteriolas que irrigan a las partes afectadas y que provocan hipoxia y necrosis focal de los tejidos blandos que lo cubren y proliferación perióstica; c) un fenómeno alérgico como la base de la enfermedad, edema e inflamación que produce una elevación perióstica con el subsiguiente depósito de calcio. El reporte de casos en diversas generaciones de una familia sugiere un posible factor hereditario, que se trasmitiría con un rasgo autosómico dominante con penetración incompleta.2,5,7
Se ha tratado de explicar la fiebre a causa de los altos niveles de prostaglandina como resultado de la excesiva actividad metabólica en los huesos. Como se han descrito lesiones óseas similares en lactantes que reciben prostaglandina exógena, uno de los mecanismos propuestos es la alteración de las prostaglandinas endógenas.8
Desde el punto de vista de su presentación existen tres manifestaciones que son comunes en la mayoría de los pacientes: a) la irritabilidad que estuvo presente en los tres pacientes e incluso fue el primer síntoma; b) la tumefacción de los tejidos blandos de aparición brusca, con características dura leñosa y dolorosa en la fase activa, que en nuestros pacientes aparecieron después de los 12 días de inicio del cuadro febril y c) el engrosamiento cortical del hueso subyacente.1,2,4,6,7 El hueso afectado fue la mandíbula en los 3 pacientes.
Otros síntomas que se reportan con relativa frecuencia son la fiebre, la palidez, seudoparálisis, disfagia y la pleuresia.1-4 En los tres pacientes nuestros estuvo presente la fiebre y la palidez cutáneo-mucosa, y debemos señalar que presentaron además hipotonía de los miembros.
No existe prueba de laboratorio específica de la enfermedad, pero los hallazgos más constantes son la eritrosedimentación acelerada y la fosfatasa alcalina aumentada en el suero sanguíneo durante la fase activa de la tumefacción y la fiebre. Todas estuvieron presentes en nuestros pacientes. La anemia por déficit de hierro es frecuente y en muchos pacientes se ha reportado la leucocitosis, que también estuvo presente en 2 de los pacientes estudiados.1,2,4,7 Otros estudios de laboratorio son normales y todos los intentos de aislar algún virus o bacteria en tejidos o líquidos corporales han sido infructuosos, aunque se han reportado valores elevados de proteína C reactiva, trombocitosis severa y pacientes con fosfatasa alcalina normal.8 En nuestros pacientes todos los cultivos fueron negativos.
La radiografía ósea es un medio diagnóstico más definitivo que la ecografía en esta enfermedad. Los hallazgos radiográficos están dados por reacción perióstica y engrosamiento de la cortical de las estructuras óseas subyacentes y la formación de hueso nuevo. Estos cambios radiográficos pueden no estar presentes en pacientes con sintomatología florida en la presentación inicial de la enfermedad.2 En los tres pacientes estudiados las alteraciones se presentaron días después de que apareció la tumoración.
Se han realizado estudios gammagráficos óseos que muestran hipercaptación del radiofármaco de la zona afectada y que se hace positivo antes que los signos radiográficos.9
El diagnóstico de la HCI se concibe con los hallazgos clínicos y radiográficos que son bastante característicos, la edad de aparición y los resultados de laboratorio. No obstante es necesario establecer diagnóstico diferencial con cuadro de abuso infantil, osteomielitis, traumatismo, neoplasia como el sarcoma de Ewing, metástasis de tumores como el neuroblastoma y parotiditis. La mayor dificultad inicial es la diferenciación con sepsis no localizada.
En nuestros pacientes el diagnóstico de la enfermedad se basó en la edad de presentación antes de los 6 meses, en la presencia de irritabilidad, fiebre, la aparición brusca de la tumoración, la presencia de engrosamiento cortical de los huesos adyacentes y de las alteraciones de laboratorio (eritosedimentación acelerada, aumento de la fosfatasa alcalina y anemia). La evolución autolimitada de la enfermedad en estos pacientes confirmó el diagnóstico.
No existe ningún tratamiento específico contra la enfermedad. Cuando existe sintomatología dolorosa se pueden indicar antiinflamatorios no esteroideos o corticoterapia. En nuestros pacientes el tratamiento inicial fue con cefalosporina de tercera generación debido al inicio de fiebre, leucocitosis, eritrosedimentación acelerada y la ausencia de tumoración, pero con esta conducta no hubo modificación del cuadro clínico ni del humoral. Otros autores también han utilizado antibióticos al inicio de la enfermedad, por confundirse con un cuadro séptico.8 Dutta y cols.8 señalan el uso de naproxeno e indometacina por otros autores con aparente buen resultado.10,11
REFERENCIAS BIBLIOGRÁFICAS
1. Caffey J. Pediatric X-Ray Diagnosis. The year Book Publishers, 1956. pp. 882-290.
2. Shafer WG, Levy BM. Tratado de Patología Bucal. 4ta. Ed ilustrada. México, D.F: Nueva editorial Interamericana. S.A ; 1993. pp. 705-706.
3. Schelger S, Chaoul R, Tennstedt C, Lehmann K, Mundlos S, Tinschert S. Antenatal onset of cortical hyperostosis (Caffey disease): Case report and review. J Med Genet. 2003 Aug 1; 120(4):547-52.
4. Delgado WA, Arrascue M. Hiperostosis cortical infantil (enfermedad de Caffey). Rev Estomat Herediana. 2007; 14. Disponible en: www.scielo.org.pe/scielo.php?pid=S1019
5. Daza Díaz AM. Hiperostosis cortical infantil. [En línea] 2000. Disponible en: www.encolombia.com/pediatria3522000
6. Díaz XE, García J, Blanco JA, Zaldo A. Enfermedad de Caffey. Revisión de cinco casos. Trabajo para optar por el título de Especialista de Primer Grado de Radiología. Ciudad de La Habana. 1982.
7. Sarmiento A, Maia I, Belo E, Rocha F, Mendaha S, Lima M. Enfermedad de Caffey o hiperostosis cortical infantil. Caso clínico. Anales Españoles de Pediatría. 2002; 56:119-21.
8. Dutta S, Jain N, Bhattachariya A, Mukhopadhyay K. Infantile Cortical Hyperostosis. Indian Pediatric 2006; 42:84-88.
9. Seatci I, Brown JJ, McAlister WH. MR findings in a patient with Caffey‘s Disease. Pediatr Radiol. 1996; 26:70.
10. Thometz JG, DiRaimando CA. A case of recurrent Caffey disease treated with naproxen. Clin Orthop. 1996; 323:304-309.
11. Couper RT, McPhee A, Morris L. Indomethacin treatment of infantile cortical hyperostosis in twins. J Pediatr Child Health. 2001; 37:305-308.
Recibido: 22 de marzo de 2006. Aprobado: 18 de mayo de 2006.
Dra. Neri Campañá Cobas. Doble Vía San Francisco y Perla, Altahabana. Ciudad de La Habana.
1 Especialista de I Grado en Pediatría. Diplomada de Nefrología.
2 Profesor e Investigador de Mérito. Instituto Nacional de Oncología.
3 Profesor Consultante de Pediatría. Hospital “William Soler”.
4 Profesora Consultante de Radiología. Hospital “William Soler”.
5 Residente de 3er año de Pediatría. Hospital “William Soler”.

