










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr v.79 n.3 Ciudad de la Habana jul.-sep. 2007
Hospital Pediátrico Provincial Docente «William Soler»
Enfermedad renal poliquística autosómica dominante
RESUMEN
El objetivo de esta revisión es trasmitir a los estudiantes y médicos generales básicos algunos aspectos fundamentales sobre esta enfermedad, relativamente frecuente, que puede incluso diagnosticarse antes del nacimiento, en el niño recién nacido, lactante y preescolar, y que hasta hace algunos años era conocida como «enfermedad renal poliquística de tipo adulto». Se enfatiza también en la importancia de la historia familiar y la utilidad del ultrasonido diagnóstico como elemento importante y no invasivo para el diagnóstico. En segundo lugar, insistimos en los signos de presentación de la enfermedad, sus posibles complicaciones y factores de riesgo para la progresión de ésta. Esta revisión aspira, además, a contribuir a la búsqueda de la enfermedad y a que se pueda ofrecer un asesoramiento genético adecuado.
Palabras clave: Quistes renales, enfermedad poliquística autosómica dominante.
La enfermedad renal poliquística autosómica dominante (EPAD) es una enfermedad hereditaria frecuente y es la causa del 10 % de las insuficiencias renales crónicas terminales en pacientes con tratamiento dialítico.1-4 Es 20 veces más frecuente que la enfermedad de Huntington, 15 veces más frecuente que la fibrosis quística del páncreas y 10 veces más frecuente que la anemia drepanocítica.2 La prevalencia citada en la literatura es de 1 por 1000 habitantes2-5 y cada descendiente de un sujeto afecto tiene 50 % de probabilidad teórica de heredar la enfermedad.4-6
Esta enfermedad es una de las primeras causas de insuficiencia renal crónica en el adulto y ha ganado importancia en pediatría desde que se describieron los primeros casos en niños.7-10 Desde hace más de 20 años es posible su diagnóstico prenatal en determinados casos,11-13 y el reporte más temprano es a las 16 semanas del embarazo.14 Se estima que aproximadamente el 2 % de los portadores del gen se presenta con manifestaciones graves en los niños.11 La inesperada aparición del comienzo infantil de la enfermedad en una familia con historia de enfermedad con inicio clásico en el adulto no tiene una explicación satisfactoria, aunque se reportan algunas variaciones intrafamiliares.15
Zerres y otros, que estudiaron 64 familias con 79 niños afectados, diagnosticaron 5 casos prenatalmente en 17 familias con más de un hijo con la enfermedad.11 En la literatura revisada por estos autores citan 23 casos diagnosticados al nacimiento y 6 antes de nacer, aunque existen otros reportes que no fueron revisados por ellos.16,17 Se ha señalado que aproximadamente en el 30 % de las personas en riesgo, el diagnóstico puede establecerse por ultrasonografía antes de los 10 años de edad,18 aunque algunos no han podido detectar quistes en el 40 % de los portadores de la enfermedad menores de 30 años.19
La insuficiencia renal puede presentarse desde los 2 hasta los 80 años.2 En una serie de 515 pacientes, el 39 % desarrolló insuficiencia renal crónica a los 54 ± 10 años de edad.4 En el análisis de otra serie de pacientes solamente el 25 % la tenía a los 50 años, mientras que a los 58 la padecía el 43 % y a los 73 años el 53 % se hallaba en insuficiencia renal crónica terminal.20 En general, a los 70 años el 50 % de los casos padece insuficiencia renal crónica terminal.21
Teniendo en cuenta que por los cálculos de frecuencia de la enfermedad en Cuba tendremos aproximadamente 10 000 sujetos padeciéndola,22 no sólo el nefrólogo y el genetista sino también el internista, el generalista, el pediatra, el médico general básico e incluso el perinatólogo, estamos en la obligación de conocer mejor esta enfermedad para tratar más adecuadamente a estos pacientes.
HISTORIA Y ETIOLOGÍA
Algunos refieren que esta enfermedad fue reportada por primera vez en Francia a comienzos del siglo XVII22 y otros la sitúan a finales del siglo XIX.23,24 La naturaleza hereditaria de esta enfermedad fue reconocida desde principios del siglo XX.25,26 Los estudios de Dalgaard en 1957 establecieron el patrón de herencia autosómico dominante de la enfermedad,27 pero no fue hasta 1985 que se localizó un gen asociado con el riñón poliquístico autosómico dominante en el brazo corto del cromosoma 16 (16 p 13.3).
Posteriormente se vio que en algunas familias no se detectan marcadores en el brazo corto del cromosoma 16,29 lo cual indicó que existía un segundo gen.2 Al primer grupo se le denominó riñón poliquístico autosómico dominante de tipo 1 (siglas en ingles: ADPKD1 ‘autosomic dominant polycystic kidney disease’) y al segundo, riñón poliquístico autosómico dominante de tipo 2 (ADPKD2). El tipo 1 es la causa de la enfermedad en cerca del 90 % o más de las familias que la padecen.30 Hasta ahora se han identificado por lo menos 3 loci diferentes como causantes de la enfermedad: PKD1, PKD2 y PKD3. El gen del tipo 2 se localiza en el brazo largo del cromosoma 4 (4 q 21-23) y el locus del PKD3, aunque es probable que comprenda más de un gen, está localizado en el cromosoma 1q a nivel de HRPT2. El tipo PKD3 sólo aparece en aisladas familias.31
FISIOPATOLOGÍA
Como la función del gen es desconocida hasta ahora, las investigaciones encaminadas a conocer la patogénesis de la enfermedad se han concentrado en los mecanismos de producción de los quistes.2 La formación de los quistes requiere la proliferación del epitelio tubular, en base a las consideraciones matemáticas impuestas por el área de superficie de los quistes y el tamaño de la célula.6 Los trabajos de microdisección han demostrado que los quistes son esencialmente divertículos a lo largo de las nefronas32,33 y en estudios con microscopía electrónica se informa que el 27 % de los quistes están conectados al glomérulo y el 37 % son sacos autónomos.34
Para la formación de los quistes renales, Gardner ha desarrollado tres hipótesis fundamentales y agrega una cuarta posibilidad.25 La primera hipótesis plantea como el defecto primario una debilidad aumentada de la membrana basal, que es sugerida por la alteración de estructura y función de la membrana.35,36 Además, las dilataciones quísticas y diverticulares en otros órganos apoyan que los pacientes con enfermedad poliquística autosómica dominante tengan una membrana basal laxa en sitios extrarrenales.
La segunda hipótesis involucra el movimiento invertido de agua y solutos, que fluye y efluye de las nefronas afectadas. Esta hipótesis es apoyada experimentalmente por lo menos por tres observaciones: a) la ATP-asa sodio-potasio parece mediar en la formación de quistes en los cultivos de órganos metanéfricos;37 b) la pared del quiste de un riñón poliquístico es metabólicamente activa y farmacológicamente respondedora in vitro;38 y c) estructuras similares a los quistes son formadas por las células MDCK cuando crecen in vitro en gel de colágeno.39
La tercera hipótesis implica la hiperplasia epitelial, los mucolípidos40 o ambas como causa de obstrucción parcial del flujo tubular.41,42 Se produce la dilatación como consecuencia del aumento de la presión intraluminal que existe en estos riñones.40,43,44
La cuarta hipótesis es la llamada hipótesis de la «proliferación» y considera que los quistes se forman debido a un crecimiento anormal hacia afuera. Apoya este postulado la hiperplasia celular observada en los modelos animales de enfermedad quística y el hallazgo de pólipos y tumores en la enfermedad quística renal humana.42,44,45
Cada una de estas hipótesis tiene defectos reconocidos y, aunque separadas, no son completamente excluyentes.25
Estudios que han examinado cuidadosamente quistes individuales sugieren que cuando el quiste crece pierde su comunicación tubular y se aísla del glomérulo, por lo que requiere transporte transepitelial de solutos y líquidos para su crecimiento posterior.34 El contenido de los quistes tiene diferente concentración de sodio46,47 y esta diferencia se ha interpretado como un reflejo del diferente sitio de origen dentro de la nefrona.2
Los modelos animales de enfermedad quística renal y el cultivo de células del epitelio quístico del riñón humano demuestran que las alteraciones en el crecimiento celular, la secreción de líquido, y la composición de la matriz extracelular, están involucradas en la quistogénesis.48,49 Los cultivos primarios del epitelio del riñón poliquístico humano tienen una respuesta proliferativa amplificada a factores de crecimiento como el factor de crecimiento epidérmico50 y una respuesta inhibidora disminuida a los inhibidores de la proliferación, como el factor B de crecimiento tumoral.51
Puede haber un aspecto autocrino o paracrino para el crecimiento de los quistes adquiridos. En los modelos animales los quistes inducidos químicamente y los cultivos de órganos metanéfricos de ratones con enfermedad poliquística recesiva regresan cuando se retira el producto químico o la hormona que promovió su formación.36 En los pacientes con enfermedad renal de cualquier etiología que desarrollan quistes durante la insuficiencia renal crónica,52 estos pueden regresar después del trasplante.3 Gabow plantea que teóricamente los quistes de la enfermedad poliquística autosómica dominante pueden regresar.2 Ye y Grantham han demostrado que los quistes renales epiteliales de los pacientes con enfermedad renal poliquística autosómica dominante pueden segregar líquido in vitro y que el aumento o disminución del líquido está relacionado con el transporte de sodio.54
ANATOMÍA PATOLÓGICA
Los riñones en la enfermedad poliquística autosómica dominante se hallan casi siempre aumentados de tamaño. Kissane refiere un caso en que cada riñón pesaba 2 600 g23 y Gabow señala que los riñones pueden exceder los 40 cm de altura y pesar hasta 8 kg.2 Los quistes pueden variar de tamaño: entre pocos milímetros y varios centímetros, y su contenido puede ser claro, turbio o achocolatado, en caso de hemorragia.2 Estos quistes de tamaño irregular producen distorsión marcada del contorno renal y de los cálices, y están cubiertos por epitelio plano.55 El estudio microscópico demuestra que pueden originarse en cualquier parte de la nefrona y no se trata en realidad de quistes sino de una invaginación sacular de la nefrona.23 Aunque la presentación bilateral es la norma, el 27 % de los casos son asimétricos, especialmente en los niños.31
Las formas asimétricas extremas se corresponden con el llamado riñón poliquístico autosómico dominante unilateral.56 El riñón poliquístico autosómico dominante con agenesia renal contralateral se ha reportado 4 veces en la literatura.31,57 En este sentido se ha señalado que es difícil corroborar si los casos de «riñones poliquísticos unilaterales del adulto» reportados antes de 1980 eran verdaderas EPAD u otros tipos de enfermedades quísticas.31
MANIFESTACIONES CLÍNICAS
Aunque se puede establecer el diagnóstico prenatal de esta enfermedad en aislados casos con historia familiar y se reportan pacientes con síntomas en la edad pediátrica, incluso neonatales,58el comienzo de la sintomatología suele iniciarse en la cuarta década de la vida.52
Los síntomas principales que hacen que los pacientes no detectados en pesquisajes familiares consulten al médico son: hipertensión arterial, dolor en el flanco, infección urinaria, hematuria y nefrolitiasis. Cada una de estas manifestaciones aparecen en el 20 a 30 % de los casos.1 La tumoración unilateral o bilateral también puede ser un hallazgo relativamente frecuente.25
La hipertensión arterial se presenta en el 30 % de los niños,59-61 el 60 % de los adultos antes que desarrollen insuficiencia renal terminal1 y el 80 % de las insuficiencias renales crónicas terminales.62 El aumento de la tensión arterial parece estar mediado por la activación del sistema renina-angiotensina, que produce un evidente estrechamiento de la red arterial visible en la arteriografía, y se han encontrado prominentes gránulos de renina en las células de la mácula densa.5 Al inicio de la enfermedad puede haber una expansión de volumen con una inapropiada falta de respuesta del sistema renina-angiotensina. La patogénesis de la hipertensión puede estar mediada por diferentes mecanismos en el curso de la enfermedad3 y su aparición está considerada un signo de mal pronóstico.63
El dolor abdominal, en la espalda o en el flanco es uno de los síntomas frecuentes de la enfermedad; el 60 % de los pacientes lo padece. Se plantea que el dolor es debido a los riñones grandes, impresión que es apoyada por la observación de que el 80 % de los pacientes esté libre de síntomas durante 1 año y el 60 %, 2 años, si se le descomprimen los quistes.2,3 El drenaje de los quistes sólo debe hacerse cuando interfiere con las actividades normales del paciente,64 porque aunque el dolor puede producirse por la acumulación de líquido en los quistes con aumento de la presión intraluminal, la causa no está totalmente aclarada.3
La infección urinaria se presenta en más de la tercera parte de los casos en algún momento de la vida, siendo más frecuente en las mujeres (72 %) que en los hombres (28 %).65 Por su localización, la infección puede dividirse en pionefrosis (infección de pelvis y cálices), infección renal parenquimatosa (infección bacteriana intersticial aguda) y piocisto (infección del quiste).65 La instrumentación es el mayor riesgo de infección y se presenta posteriormente a ésta en el 40 % de los casos, si no se utiliza el antibiótico adecuado.65 La infección quística puede ser difícil de diferenciar de la infección renal, aunque en la infección quística es más frecuente el dolor lumbar; en la infección renal es más frecuente el hemocultivo positivo y en la pionefrosis es más frecuente la leucocituria y la cilindruria.66 Estas diferencias son importantes para decidir el tratamiento. La infección perinefrítica tiene alta mortalidad.66
La hematuria es el signo de presentación en el 35 % de los casos y en el 50 % de los pacientes se observa hematuria macroscópica o microscópica.2 El riesgo de hematuria parece asociarse a la hipertensión y el aumento del tamaño de los quistes. Aunque la mayoría de los pacientes señalan causas precipitantes como traumas y ejercicios violentos, no se ha demostrado una asociación precisa.2,67
La nefrolitiasis se señala en el 15 a 20 % de los casos y ésta puede producir dolor y hematuria. Los cálculos están constituidos principalmente de urato y oxalato de calcio y están asociados con pH urinario bajo, descenso de la excreción urinaria de citrato y aumento de la excreción de ácido úrico.68
Se presenta proteinuria en aproximadamente un tercio de los pacientes, pero ésta es casi siempre ligera (< 1 g en 24 h). Los casos con proteinuria de rango nefrótico a los que se les ha realizado biopsia renal, tenían sobreañadida una enfermedad glomerular.69-72
A los 60 años de edad aproximadamente, el 45 % de los pacientes presenta insuficiencia renal.20,63 Sin embargo, la edad de comienzo de la insuficiencia renal puede variar ampliamente y se señala que esta variabilidad está influida por el tipo genético de la enfermedad, ya que los pacientes con el tipo 2 tienen mayor edad al comienzo de la insuficiencia renal que los pacientes del tipo 1.2 Pero, otros factores pueden influir en la progresión de la enfermedad renal y ésta parece ser menos agresiva en la mujer que en el hombre.
Ciertas funciones endocrinas del riñón también parecen alterarse, reflejándose tanto en la secreción de renina como de eritropoyetina. La secreción de renina está relacionada con la hipertensión arterial como se señaló anteriormente, y los niveles altos de eritropoyetina ayudan a mantener un nivel de hemoglobina y hematocrito más altos que en las insuficiencias renales por otras causas,73 pudiendo llegar a producirse policitemia, pero sólo en raros casos.74-76 Los niveles elevados de eritropoyetina pueden producirse en parte por su secreción por las células relacionadas con el quiste y existe una relación directa entre los niveles de la hormona en el líquido quístico y en el suero, por lo que puede ocurrir que al aumentar los quistes ascienda el nivel sanguíneo de eritropoyetina.2
Teniendo en cuenta lo señalado de que ésta no es sólo una enfermedad renal,5 es importante analizar sus manifestaciones extrarrenales. Las manifestaciones extrarrenales pueden ser quísticas y no quísticas. Los quistes se presentan en el hígado, ovarios, páncreas y bazo.2,27,77 Las manifestaciones no quísticas incluyen las anormalidades de las válvulas cardíacas, los aneurismas cerebrales y los divertículos del colon.2,3,25
Los quistes hepáticos se originan probablemente del epitelio biliar y aparecen aproximadamente 10 años después de los quistes renales.3 Cerca del 50 % de los pacientes sin insuficiencia renal tienen quistes hepáticos.2 Estos quistes aumentan con la edad y son infrecuentes en el niño; entre los 20 y 30 años el 10 % de los pacientes tiene quistes hepáticos y a los 60 años los tiene el 75 % de estos enfermos. Aunque los quistes hepáticos se ven en ambos sexos, la masividad, el gran tamaño y las complicaciones son más frecuentes en la mujer, lo que hace plantear que este aspecto está relacionado con las hormonas esteroides femeninas. Se ha visto que las mujeres con mayor número de embarazos tienen más quistes y estos tienden a ser más grandes.2,3 Los síntomas relacionados con los quistes hepáticos son raros y obedecen a la compresión de estructuras vecinas. Lieske y Toback hacen referencia a una serie de pacientes en hemodiálisis, en quienes las complicaciones relacionadas con los quistes hepáticos (infección, hipertensión portal y colangiocarcinoma) ocasionaron el 10,5 % de las muertes.2
Refiere Gabow que no hay datos publicados sobre la frecuencia de los quistes ováricos, aunque éstos se han reportado en esta enfermedad y este parece ser un hallazgo frecuente.2
No hemos encontrado referencias en cuanto a los quistes pancreáticos y esplénicos.
Las principales manifestaciones extrarrenales no quísticas son las relacionadas con las alteraciones valvulares cardíacas.78,79 El 26 % de los pacientes con enfermedad renal poliquística autosómica dominante tienen prolapso de la válvula mitral, comparado con el 2 % del grupo control de la población.79 El prolapso de la mitral en la población pediátrica se reporta en el 2,6 % y en los adolescentes alcanza el 5,1 %.80 Los síntomas imputables al prolapso de la mitral, como las palpitaciones y el dolor precordial, son mucho más frecuente en la enfermedad poliquística autosómica dominante que en la población normal. Las anormalidades de la válvula aórtica pueden requerir tratamiento quirúrgico77,78 y tanto la válvula aórtica como la tricúspide pueden tener degeneración mixomatosa.78
Los aneurismas saculares del cerebro están presentes en el 10 a 40 % de los pacientes y son visibles en la arteriografía o en la autopsia,5 aunque su verdadera frecuencia se desconoce por ser muy variables los resultados de diferentes estudios reportados y esto puede estar influenciado por la selección de los pacientes, los diferentes medios diagnósticos empleados y las diferencias entre los grupos étnicos estudiados.2 Refiere Gabow que en tres estudios realizados en los Estados Unidos los resultados también son variables. En el primero se estudiaron 96 pacientes mediante resonancia magnética y tomografía computarizada y no encontró aneurismas; el segundo halló a 4 pacientes con aneurismas entre 92 estudiados con tomografía computarizada, angiografía o ambas y el tercer estudio detectó 9 casos asintomáticos, al estudiar 85 casos con resonancia magnética.2 Se discute el uso de la angiografía en pacientes con antecedentes familiares de hemorragia subaracnoidea,3 pero se ha recomendado la tomografía axial de alta resolución o la resonancia magnética en secciones de 3 mm a lo largo del polígono de Willis en los individuos con alto riesgo.80
Los divertículos de colon se reportan en el 80 % de los casos en hemodiálisis por insuficiencia renal crónica debido a esta enfermedad.81 Aunque la patogénesis de la enfermedad diverticular no está bien definida2 se recomienda la evaluación de estos divertículos antes del trasplante renal y considerar la colectomía parcial si se encuentra enfermedad diverticular extensa.3
DIAGNÓSTICO
Para el diagnóstico de la enfermedad poliquística se necesita la presencia de 3 a 5 quistes en cada riñón.3 En el estadio presintomático de la enfermedad, el análisis genético molecular basado en el ligamiento de ella a marcadores polimórficos de DNA situados próximos al denominado locus PKD1 en el brazo corto del cromosoma 16, hace que sea posible la predicción diagnóstica precoz en más del 90 % de los casos.4,82,83 En este período presintomático el ultrasonido puede detectar quistes en el 30 % aproximadamente de los menores de 10 años, entre 60 y 83 % de los comprendidos entre 10 y 30 años (figura) y prácticamente en el 100 % de los mayores de 30 años.3,11 Debe señalarse que aproximadamente el 10 % de los casos corresponden al tipo 2. La mitad de los pacientes con EPAD tienen quistes en otros órganos (principalmente en hígado, páncreas, bazo y pulmones).31
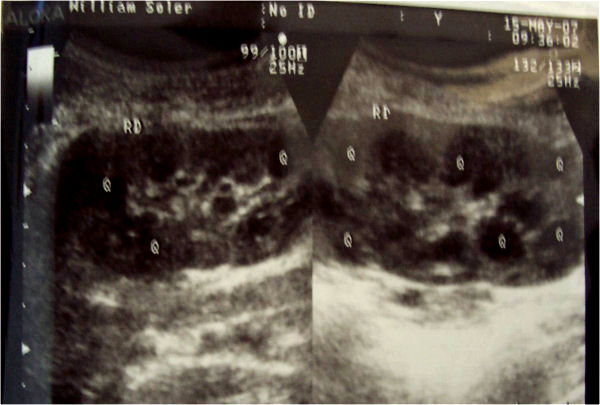
Figura. Enfermedad renal poliquística en un niña de 12 años.
La historia familiar es un elemento muy importante para el diagnóstico tanto en el tipo 1 como en el tipo 2, y cuando es típica ayuda al diagnóstico en el 90 % de los casos; el 10 % constituye una mutación genética por lo que la historia familiar es negativa. Cuando la historia familiar es típica, el ultrasonido detecta más de 3 quistes por riñón y el estudio genético molecular detecta la alteración en el brazo corto del cromosoma 16 o en el brazo largo del cromosoma 4, se tiene el diagnóstico de certeza. Cuando se detectan más de 3 quistes por riñón y la historia familiar es típica también se puede hacer el diagnóstico. Cuando esto no es así y aparecen quistes renales sin antecedentes familiares, es necesario valorar otras posibilidades diagnósticas.
En el caso de niños con riñones quísticos el diagnóstico diferencial más difícil puede ser la enfermedad renal poliquística autosómica recesiva (EPAR). Aunque son dos entidades distintas y con diferentes tipos de herencia, tienen presentación clínica e imaginológica que pueden confundirlas.84,85 En ocasiones es necesario recurrir al examen histológico de hígado y riñón para diferenciarlas. La variedad recesiva muestra quistes radiales derivados de los tubos colectores y tiene como característica la ectasia y proliferación de los conductos biliares con fibrosis portal,85 y en la EPAD los quistes varían de tamaño y pueden aparecer en cualquier sitio de la nefrona, afectando frecuentemente túbulos y glomérulos.15,86
En algunas ocasiones el fenotipo clásico de EPAD puede verse asociado a la esclerosis tuberosa, como un ejemplo de «síndrome de genes contiguos», porque el gen PKD1 está inmediatamente adyacente al gen TSC2 que produce las dos terceras parte de la esclerosis tuberosa.31 El gen TSC1 de esta enfermedad está situado en el cromosoma 9.87 Cuando es necesario, el diagnóstico puede establecerse mediante biopsia renal ya que los quistes de la esclerosis tuberosa muestran paredes limitadas por epitelio hiperplástico y eosinofílico que se consideran elementos patognomónicos de la enfermedad.88
Estas dos entidades, EPAR y esclerosis tuberosa, son las de mayor dificultad en cuanto al diagnóstico diferencial, porque las enfermedades que se acompañan de quistes renales, hereditarias o no, son muchas y generalmente no ofrecen confusión.
PRONÓSTICO
Como se señaló anteriormente, estos pacientes evolucionan a la insuficiencia renal terminal y, en estudios realizados alrededor de los 50 a 60 años de edad, el 39 % de los pacientes se encuentran en esta situación.4). Otras series señalan solamente un 25 % de insuficiencia renal crónica a los 50 años, 43 % a los 58, y 53 % a los 73.20). En general, a los 70 años el 50 % de los pacientes está en insuficiencia renal crónica terminal.21
La EPAD tiene un riesgo alto de que la proliferación celular produzca hiperplasia, adenoma o carcinoma. El carcinoma de células renales en esta enfermedad tiene un perfil clinicopatológico que lo distingue: pacientes jóvenes, sin predilección sexual, y con preferencia es de tipo sarcomatoso.31 Se ha señalado la utilidad de la tomografía computarizada para su diagnóstico.89 Se han reportado más de 50 casos,31 incluyendo un carcinoma de tubos colectores.90
CONDUCTA MÉDICA
La conducta médica se basa en el asesoramiento genético, el diagnóstico precoz, el tratamiento sintomático, el tratamiento de las complicaciones y el trasplante renal.
Es sabido que el individuo afectado por la enfermedad políquística autosómico dominante tiene 50 % de probabilidad teórica de trasmitir la enfermedad a su descendencia.4,6 Conociendo estas posibilidades el diagnóstico puede confirmarse mediante el estudio bioquímico molecular, la ultrasonografía, la resonancia magnética o la tomografía computarizada.3 Sin embargo, existen experiencias de diferentes actitudes entre individuos con posibilidades de tener riñones poliquísticos, que han rechazado la investigación hasta en el 22 % de los casos considerados de alto riesgo, y un bajo porcentaje decide cambios en sus planes reproductivos al conocer la enfermedad y sus posibles riesgos.3
En el paciente sintomático los episodios de hematuria son relativamente frecuentes y pueden ser graves, agudos y dolorosos. Si la hematuria no está acompañada de urolitiasis, el reposo durante algunos días es la conducta recomendada; si la hematuria se debe a una litiasis y ésta produce obstrucción urinaria, la presencia de los quistes renales puede hacer más difíciles los procederes urológicos y aumentar el riesgo de complicaciones quirúrgicas o endoscópicas. La hematuria macroscópica es el signo de presentación en algunos escolares y adolescentes en los que falta el antecedente familiar de la enfermedad.
La infección urinaria es una complicación frecuente en estos pacientes, principalmente en la mujer. De las localizaciones de la infección, ya señaladas anteriormente, la infección quística es la más seria, ya que habitualmente no cede a cursos cortos de antibióticos.65 Para el tratamiento de las pionefrosis y las infecciones intersticiales agudas pueden utilizarse los antibióticos indicados en las infecciones urinarias en general, pero en la infección quística se debe seleccionar un antibiótico que penetre en el quiste, como el sulfametoxazol-trimetoprina y el cloranfenicol.3 La ciprofloxacina sería el de elección en el adulto, pero debe tenerse en cuenta que aún no se recomienda su utilización en menores de 18 años, y pueden utilizarse las cefalosporinas de tercera generación.
Los quistes infectados y los de gran tamaño pueden producir dolor lumbar o en el flanco, por lo que primeramente se utilizó la cirugía para reducir el volumen quístico y aliviar los síntomas asociados, pero el deterioro de la función renal era un elemento importante en este proceder.91 Se ha reportado que la aspiración del contenido líquido de los quistes disminuye el dolor lumbar y desciende la tensión arterial sin deterioro de la función renal92por lo que en la actualidad se prefiere este proceder. También se ha utilizado el drenaje percutáneo cuando no hay respuesta adecuada a los antibióticos.3
Para la esclerosis de los quistes se utiliza frecuentemente el alcohol,93 y se reporta la utilidad de la minociclina con esta finalidad.93,94 Con este procedimiento se han llegado a puncionar y esclerosar hasta 12 quistes, 6 de cada riñón, de los que se han extraído 430 mL de líquido.95
La hipertensión arterial requiere tratamiento en la mayoría de estos pacientes y éste es fundamentalmente medicamentoso, aunque en ocasiones es necesario el drenaje de los quistes. Es probable que la hipertensión juegue un papel importante en los cambios vasculares observados.
Entre los 60 y 70 años el 50 % de estos pacientes ha llegado a la insuficiencia renal terminal, pero algunos mucho antes de esa edad, por lo que la hemodiálisis y el trasplante renal son necesarios en estos casos.
Entre los indicadores de mal pronóstico se señala su asociación al rasgo drepanocítico y que en los negros, la insuficiencia renal se alcanza como promedio 10 años antes que en los blancos.95 Otro indicador de mal pronóstico es el número mayor de 3 embarazos,63 por lo que se impone la orientación adecuada a la mujer con esta enfermedad. Además de estos indicadores, Gabow, y otros que estudiaron 580 pacientes con la enfermedad, incluyen como indicadores de mal pronóstico: la edad temprana al diagnóstico, el sexo masculino, la hipertrofia ventricular izquierda, la hematuria macroscópica y la infección urinaria en el hombre, así como la presencia de quistes hepáticos en la mujer. También se reporta peor pronóstico en los pacientes con alteraciones del cromosoma 16.63
summary
The objective of this literature review was to provide medical students and general practitioners with some fundamental aspects on this relatively frequent disease that may even be diagnosed before birth, in newborns, infants and preschool-age children and that until some years, it was known as “polycystic autosomal recessive disease of the adult type”. The importance of family history and usefulness of diagnostic ultrasound as an important non-invasive element of diagnosis was stressed. Secondly, we insist on the presentation signs of the disease, possible complications and risk factors for the progression. This review was additionally intended to contribute to detection of the disease and to provide an adequate genetic counselling.
Key words: renal cysts, autosomal dominant polycystic disease.
REFERENCIAS BIBLIOGRÁFICAS
1. Zeier M, Geberth S, Ritz E, Jaeger T, Waldheer R. Adult dominant polycystic kidney disease: Clinical problems. Nephron. 1988;49:177-183.
2. Gabow PA. Autosomal dominant polycystic kidney disease. N Engl J Med. 1993;329:332-342.
3. Lieske JC, Toback FG. Autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1993;3:1442-1449.
4. Rexistro Galego de Nefropatías (RGN). Epidemiología de la enfermedad renal poliquística autosómica dominante en Galicia. Impacto del tratamiento sustitutivo. Nefrología. 1993; 13: 122-126.
5. Gabow PA. Autosomal dominant polycystic kidney disease: More than a renal disease. Am J Kidney Dis. 1990; 16: 403-413.
6. Gabow PA. Polycystic kidney disease: Clues to pathogenesis. Kidney Int. 1991; 40: 989-996.
7. Blyth H, Ockenden BG. Polycystic disease of kidneys and liver presenting in childhood. J Med Genet. 1971; 8: 257-284.
8. Kaye C, Lewy PR. Congenital appearance of adult-type (autosomal dominant) polycystic kidney disease: Report of a case. J Pediatr. 1974; 85: 807-810.
9. Stickler GB, Kelalis PP. Polycystic kidney disease: Recognition of the "adult form" (autosomal dominant) in infancy. Mayo Clin Proc. 1975; 50: 547-548.
10. Kaplan BS, Rabin I, Nogrady MB, Drummond KN: Autosomal dominant polycystic renal disease in children. J Pediatr. 1977; 90: 782-783.
11. Zerres K, Rudnik-Schöneborn S, Deget F. Childhood onset autosomal dominant polycystic kidney disease in sibs: Clinical picture and recurrence risk. J Med Genet. 1993; 30: 583-588.
12. Zerres K, Weiss H, Bulla M, Roth B: Prenatal diagnosis and early manifestation of autosomal dominant adult-type polycystic kidney disease. Lancet. 1992; II: 988.
13. Pretorius DH, Lee ME, Manco-Johnson ML, Weingast GR, Sedman AB, Gabow PA: Diagnosis of autosomal dominant polycystic kidney disease in uterus and in the young infant. J Ultrasound Med. 1987; 6: 249-255.
14. Fryns JP, Vanderberghe K, Moerman F. Mild-trimester ultrasonographic diagnosis of early manifestations "adult" form of polycystic kidney disease. Hum Genet. 1986;74: 461.
15. Bernstein J. Glomerulocystic kidney disease-Nosological considerations. Pediatr Nephrol. 1993;7: 464-470.
16. Chevalier RL, Garland TA, Buschi AJ. The neonate with adult-type autosomal dominant polycystic kidney disease. Int J Pediatr Nephrol. 1981; 2: 73-77.
17. Edwards OP, Baldinger S. Prenatal onset of autosomal dominant polycystic kidney disease. Urology. 1989; 34: 265-270.
18. Baer JC, Parfrey PS, Morgan JM, Martin CJ, Cramer BC. Autosomal dominant polycystic kidney disease: New information for genetic counseling. Am J Med Genet. 1992; 48: 548-553.
19. Coto E, Aguado S, Álvarez J, Menéndez Díaz MJ, López Larrea C. Genetic and clinical studies in autosomal dominant polycystic kidney disease type 1. J Med Genet. 1992; 29: 243-246.
20. Churchill DN, Bear CJ, Morgan J, Payne RH, McManamon PJ, Gault MH. Prognosis of adult onset polycystic kidney disease re-evaluated. Kidney Int. 1984; 26: 190-193.
21. Grantham JJ. Polycystic kidney, an old problem in a new context. N Engl J Med. 1988;319: 944 -946.
22. Dávalos JM. Quistes del riñón. En: "Consulta Médica". Periódico Granma. Sábado 1ro de abril de 1989.
23. Kissane JM. Enfermedad poliquística del adulto. En: Nefrología. Hamburger J, Crosnier J, Grünfeld JP, (Eds), Tomo 2. capítulo 42. La Habana: Edición Revolucionaria;1983. P. 888.
24. Lejars F. Du gros rein polykystique de l'adulte. Steinheill. Paris, 1888: 5 (citado en 23).
25. Gardner KD. Cystyc kidney. Kidney Int. 1988; 33: 610-621.
26. Cairns HWB. Heredity in polycystic disease of the kidneys. Q J Med. 1926; 18: 359 (citado en 25).
27. Dalgaar OZ. Bilateral polycystic disease of the kidneys; A follow-up of two hundred and eighty-four patients and their families. Acta Med Scand (Suppl) 1957: 1-255,-
28. Reeders ST, Breuning MH, Davies KE, Nicholds RD, Jarman AP, Higgs DR, et al. A highly polymorphic DNA marker linked to adult polycystic kidney disease on chromosome 16. Nature. 1985; 317: 542-543.
29. Kimberline WJ, Fain PR, Kenyon JB, Goldgar D, Sujansky E, Gabow PA. Linkage heterogeneity of autosomal dominant polycystic kidney disease. N Engl J Med. 1988; 319: 913.
30. Pieke SA, Kimberline WJ, Kenyon JB, Gzbow P, Genetic heterogeneity of polycystic kidney disease. An estimate of the proportion of families unlinked to chromosome 16. Am J Hum Genet. 1989; 45 (suppl): A58 (Resumen).
31. Bisceglia M, Galliani CA, Senger C, Srallone C, Sessa A. Renal cystic diseases: A review. Adv Anat Pathol. 2006; 13: 26-56.
32. Baert L. Hereditary polycystic kidney disease (adult form): A microdisection study of two case at an early stage of the disease. Kidney Int. 1978; 13: 519-525.
33. Osathanond HV, Potter EL. Pathogenesis of polycystic kidneys. Arch Pathol. 1964; 77: 459-512.
34. Grantham JJ, Geiser JL, Evan AP. Cyst formation and growth in autosomal dominant polycystic kidney disease. Kidney Int. 1987; 31: 1145-1152.
35. Butkouski RJ, Carone FA, Grantham JJ, et al. Tubular basement membrane changes in 2-amino-4,5 diphenilthiazole-induced polycystic disease. Kidney Int. 1985; 28: 744.
36. Kanwar YS, Carone FA. Reversible changes of tubular cell and basement membrane in drug-induced renal cystic disease. Kidney Int. 1984; 26: 35-43.
37. Avner ED, Sweeney WE, Finegold DN, et al. Sodium-potassium ATP-asa activity mediates cyst formation in methanephric organ culture. Kidney Int. 1985; 28: 445.
38. Perrone RD. In vitro function of cyst epithelium from human polycystic kidney. J Clin Invest. 1985; 76: 1688-1691.
39. Uchic M, Kornhaus J, Grantham JJ, et al. Amiloride and amilorides analogues retard enlargement of MDCK-cysts grown in hydrated collagen. Kidney Int. 1978; 31: 184.
40. Gargoire JR, Torres VE, Holley KE, Farrow GM. Renal epithelial hyperplasic and neoplastic proliferation in autosomal dominant polycystic kidney disease. Am J Kidney Dis. 1987; 9: 27-38..
41. Evan AP, Gardner KD, Bernstein J. Polypoid and papillary epithelial hyperplasia: A potential cause of ductal obstruction in adult polycystic disease. Kidney Int. 1979; 16: 743-750.
42. Gardner KD, Evan AP, Bernstein J. Polyps in renal cystic disease. Lancet. 1978; 1: 878.
43. Gardner KD, Evan AP. Renal cystic disease induced by diphenilthiazole. Kidney Int. 1983; 24: 43.
44. Evan AP, Gardner KD. Nephron obstruction in nordihidroguaiaretic acid-induced renal cystic disease. Kidney Int. 1979; 15: 7-19.
45. Gardner KD, Reed WP, Evan AP, Zedalis J, Hilarides MD, Leon AA.: Endotoxin provocation of experimental renal cystic disease. Kidney Int. 1987; 32: 329-334.
46. Gardner KD. Composition of fluid in twelve cysts of a polycystic kidney. N Engl J Med. 1969; 281: 985-988.
47. Huseman R, Grady A, Welling D, Grantham J Macropuncture study of polycystic disease in adult human kidneys. Kidney Int. 1980; 18: 375-385.
48. Gattone VH, Grantham JJ. Understanding human cystic disease through experimental models. Sem Nephrol. 1991; 11: 617-631.
49. Wilson PD, Sherwood AC. Tubulocystic epithelium. Kidney Int. 1991; 39: 450-453.
50. Klingel R, Dippold W, Störkell S. Expression of differentiation antigens and growth-related genes in normal kidney, autosomal dominant polycystic kidney disease, and renal cell carcinoma, Am J Kidney Dis,1992; 19: 22.
51. Wilson PD. Aberrant epithelial cell growth in autosomal dominant polycystic kidney disease. Am J Kidney Dis. 1991; 17: 634-637.
52. Ritz E, Bommer J, Bachmann S, et al.: Multycystic transformation of kidneys in chronic renal failure. Kidney Int. 1984; 25: 175 (Resumen).
53. Ishikawa I. Acquired cystic disease: Mechanisms and manifestations. Sem Nephrol. 1991; 11: 671-684.
54. Ye M, Granthan JJ. The secretion of fluid by renal cysts from patients with autosomal polycystic kidney disease. N Engl J Med. 1993; 329: 310-313.
55. Drummond KN. Riñones poliquísticos de tipo adulto. En: Tratado de Pediatría de Nelson. Behrman RE, Vaughan VC (Eds), Tomo II. La Habana: Edición Revolucionaria;1988. P.1404.
56. Strand WR, Rushton HG, Markle BM. Autosomal dominant polycystic kidney disease in infant: Asymmetric disease mimicking a unilateral renal mass. J Urol. 1989; 141: 1151-1153.
57. Jeong GH, Park BS, Jeong TK. Unilateral autosomal dominant polycystic kidney disease with contralateral renal agenesis: A case report. J Korean Med Sci. 2003; 18: 284-286.
58. Proesmans W, Van Damme B, Casaer P, Marchal G. Autosomal dominant polycystic kidney disease in the neonatal period: Association with a cerebral arteriovenous malformation. Pediatrics. 1982; 70: 971-975.
59. Report of a Meeting of Physicians and Scientists. University College and Middlesex school of Medicine. London: Autosomal dominant polycystic kidney disease. Lancet. 1992; 339: 1146-1149.
60. Zeier M, Geberthi S, Scmidt KG, Mandelbaum A, Ritz E, Elevated blood pressure profile and left ventricular mass in children and young adults with autosomal dominant kidney disease. J Am Soc Nephrol. 1993; 3: 1451-1457.
61. Chapman A, Johnson A, Duley I Gabow P. Hypertension occurs frequently in children with autosomal dominant polycystic kidney disease, J Am Soc Nephrol, 1991; 2: 251 (Resumen).
62. Gabow P, Ikle DW, Holmes JH. Polycystic kidney disease: Prospective analysis of nonazotemic patients and family members. Ann Intern Med. 1984; 101: 238-247.
63. Gabow PA; Johnson AM, Kaehny WD, Kimberling WJ, Lezotte DC, Duley ET., Factors affecting the progression of renal disease in autosomal dominant polycystic kidney disease. Kidney Int. 1992; 41: 1311-1319.
64. Grantham JJ. Renal pain in polycystic kidney disease: When the hurt wont stop. J Am Soc Nephrol. 1992; 2: 1161-1162.
65. Sklar AH, Carvana RJ, Lammers JE, et al. Renal infections in autosomal dominant polycystic kidney disease. Am J Kidney Dis. 1987; 10: 81.
66. Schwab SJ, Bander SJ, Klahr S. Renal infection in autosomal dominant polycystic kidney disease. Am J Med. 1987; 82: 714-718.
67. Gabow PA, Duley I, Johnson AM. Clinical profiles of gross hematuria in autosomal dominant polycystic kidney disease. Am J Kidney Dis. 1992; 20: 140-143.
68. Torres VE, Erickson SB, Smith LH, Wilson DMº Hattery RR, Segura JW. The association of nephrolitiasis and autosomal dominant polycystic kidney disease. Am J Kidney Dis. 1988; 11: 318-325.
69. Abe R, Yamaguchi K, Igarashi N, Fukuda Y, Kawaguchi K. A case of polycystic kidney associated with membranous nephropathy. Kidney Dial. 1985;9: 389.
70. Murphy G, Tzamaloukis AH, Listrom MB, Gibrl LJ, Smith SM, Gardner KD. Nephrotic syndrome and rapid renal failure in autosomal dominant polycystic kidney disease. Am J Nephrol. 1990; 10: 69-72.
71. Shikata K, Makino H, Ota Z. A membranous nephropathy associated with adult polycystic kidney disease. Clin Nephrol. 1991; 36: 223-227.
72. Villar MTA, Bass P, Dewhurst G, Dathan JRE. Autosomal dominant polycystic kidney disease complicated by glomerulonephritis. Nephron. 1992; 62: 226-226,228.
73. Chandra M, Miller ME, García JF, Mossey RT, McVicar M, Serum immunoglobulin, erythropoietin levels in patients with polycystic kidney disease as compared with other hemodialysis patients. Nephron. 1985; 39: 26-29.
74. Forsell J. Nephrogenous polycytemia. Acta Med Scand. 1958; 161: 169-179.
75. Ross WF, Waldmann TA, Cohen P. Renal cysts, erythropoietin and polycytemia. Am J Med. 1963; 34: 76-81.
76. Anderson ET; Walker BR. Polycystic kidney disease, polycytemia and azotemia. JAMA. 1969; 208: 2472-2473.
77. Torres VE, Wiebers DO, Forbes GS. Cranial computed tomography and magnetic resonance imaging in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1990;1. 84-90.
78. Leier CV, Baker PB, Kilman JW, Wooley CF: Cardiovascular abnormalities associated with adult polycystic kidney disease. Ann Intern Med. 1984; 100: 683-688.
79. Hossack KF, Leddy CR, Johnson AM, Schrier RW, Gsbow PA. Echocardiographic findings in autosomal dominant polycystic kidney disease. N Engl J Med. 1988; 319: 907-912.
80. Ohara N, Nikajima T, Takagi J. Mitral valve prolapse in childhood: The incidence and clinical presentations in different age groups. Acta Pediatr JPN. 1991; 33: 467.
81. Scheff RT, Zuckerman G, Harter H, Delmez J, Koehler R. Diverticular disease in patients with chronic renal failure due to polycystic kidney disease. Ann Intern Med. 1980; 92: 202-204.
82. Breuning MH, Reeders ST, Brunner H. Adult polycystic kidney disease with flanking DNA markers. Lancet. 1987; 2: 1359-1361.
83. San Millan JC, Peral B, Valero A Hernández C, Moreno F. Estudio genético de la enfermedad del riñón poliquístico autosómico dominante (ERPAD) en la población española. Nefrología. 1992; 12: 231-238.
84. Kaarainen H, Koskimies O, Norio R. Dominant and recessive polycystic kidney disease in children: Evaluation of clinical features and laboratory data. Pediatr Nephrol. 1988; 2: 296-302.
85. Kogutt MS, Robichaux WH, Boineau G, Drake GK, Simonton SC. Asymmetric renal size in autosomal recessive polycystic kidney: A unique presentation. Am J Radiol. 1993; 160: 835-836.
86. McDonald RA, Avner ED. Inherited polycystic kidney disease in children. Sem Nephrol. 1991; 11: 632-642.
87. Fryer AE, Chalmers A, Connor JM, et al.: Evidence that the gene for tuberous sclerosis is on chromosome 9. 1987; 1: 659.
88. Bernstein J, Robbins TO, Kissane JM. The renal lesions of tuberous sclerosis. Sem Diag Pathol. 1986; 3: 97-105.
89. Lang EK, Davis R. Autosomal dominant polycystic disease with renal cell carcinoma; J Urol. 2005; 173: 987.
90. Kuroda N, Satake H, Moyazki E. Collecting duct carcinoma exhibiting diastase-resistant PAS-positive cytoplasmic inclusion and rhomboid features in adult polycystic kidney disease: A case report. Int J Surg Pathol. 2004, 12: 171-177.
91. Bricker NS; Patton CJF. Renal function studies in polycystic kidney disease. N Engl J Med. 1957; 256: 212-217.
92. Bennett WM, Elzinga L, Golper TA, Barry JM. Reduction of cyst volume for symptomatic management of autosomal dominant polycystic kidney disease. J Urol. 1987; 37: 620-626.
93. Uemasu J, Fujiwara M, Munemura C, Tokumoto A, Kawasaki H. Effects of topical instillation of aminocycline hydrochloride on cyst size and renal function in polycystic kidney disease. Clin Nephrol. 1993; 39: 140-144.
94. Yamaguchi M; Koshida Y, Takimara H, Treatment of renal cysts with minocycline. Kinyoki-Geka. 1989; 2: 1119 (citado en 93).
95. Yium JJ, Gabow PA, Johnson A, Kimberling W, Kawasaki H: Autosomal dominant polycystic kidneys in blacks with ESRD: Frequency, clinical course, and effects of sickle-cell hemoglobin. J Am Soc Nephrol. 1994; 3: 305 (Resumen).
Recibido: 26 de febrero de 2007. Aprobado: 19 de abril de 2007.
Dr. Sandalio Durán Álvarez. Ave San Francisco y Perla, Altahabana. La Habana, Cuba.
Correo electrónico: sduran@infomed.sld.cu
1 Especialista de II Grado en Pediatría. Profesor Consultante de Pediatría.

