










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr v.79 n.3 Ciudad de la Habana jul.-sep. 2007
Presentaciones de casos
Hospital Pediátrico Docente «Juan M. Márquez»
Epilepsia de ausencia de inicio precoz: a propósito de un caso
Dra. Ileana Valdivia Álvarez,1 Dra. Liane Aguilar Fabré2 y Lic. Mario Caraballo Pupo3
RESUMEN
Se presenta un caso de epilepsia de ausencia de inicio antes de los 2 años de edad, que requirió múltiples drogas antiepilépticas. Se revisa la bibliografía sobre el tema y se profundiza en las actuales variaciones de los criterios diagnósticos en relación con los síndromes de ausencia epiléptica y las variantes de presentación que pueden ser causa de errores diagnósticos y terapéuticos. Se confirma el importante papel de la monitorización electroencefalográfica y videoelectroencefalográfica como herramienta diagnóstica en las epilepsias de presentación poco común en la infancia. Se revisan los factores etiológicos polimórficos actuales, el papel de los canales iónicos y el uso de las drogas antiepilépticas en la ausencia infantil.
Palabras clave: Epilepsia de ausencia, petit mal, etosuximida, canales de calcio, videoelectroencefalograma.
La epilepsia es uno de los problemas neurológicos más frecuentes y, más que una enfermedad, debe considerarse como síntoma de una alteración cerebral que puede tener la más diversa etiología.
No existe en nuestro organismo ninguna función que no sea regulada por el sistema nervioso, lo que nos permite entender la gran variedad de manifestaciones clínicas con que se pueden expresar las crisis epilépticas, y si no estamos familiarizados con ellas, será difícil diagnosticar correctamente a los pacientes. Todo síntoma o evento clínico, aún el más sutil, que aparezca de cuando en cuando, que sea de duración breve, que siempre manifieste las mismas características y que sea independiente de la voluntad del paciente, deberá sugerirnos que se trata de un evento convulsivo.
Entre los tipos de crisis que ocurren en las epilepsias generalizadas, las crisis de ausencia y las mioclónicas son las más relevantes en el contexto del diagnóstico diferencial.1
Las crisis de ausencia se manifiestan con disminución o pérdida del estado de conciencia, con inicio y final súbitos y sin confusión después del ictus epiléptico. Los pacientes presentan interrupción de la actividad motora, asociadas o no a automatismos o fenómenos autonómicos. Estas manifestaciones se traducen por complejos punta-onda lentos y generalizados en el electroencefalograma (EEG).2,3
En algunos pacientes, el diagnóstico diferencial entre las crisis parciales complejas y las crisis de ausencia puede ser muy difícil, lo que conduce a un diagnóstico erróneo del síndrome epiléptico.2,4,5
El inicio de las crisis de ausencia se sitúa entre los 4 y 9 años,6 pero puede tener límites más amplios y comenzar de forma más temprana a partir de los 3 años, siendo excepcional su inicio en edades más tempranas,7,8 en las que se señala un pronóstico menos favorable.9
Las crisis de ausencia, aparte de conformar un síndrome epiléptico específico de la infancia, pueden suponer la expresión inicial de distintos síndromes epilépticos,10 por lo que el control evolutivo es esencial para corroborar el diagnóstico. El trazado electroencefalográfico es muy útil para clasificar las ausencias epilépticas, pero en los casos de duda diagnóstica, la realización de un video-EEG es casi imprescindible.11
Las ausencias epilépticas se tratan con drogas específicas, y en la actualidad las más utilizadas son el valproato de sodio (VPA), etosuximida (ESM) y lamotrigina (LTG), aunque ninguna ha demostrado ser superior a otras en los estudios de medicina basada en evidencias.12
Presentamos un caso de epilepsia de ausencia de inicio muy temprano, de difícil diagnóstico y tratamiento, y se revisa la bibliografía sobre el tema.
PRESENTACIÓN DEL CASO
Paciente del sexo femenino, producto de un embarazo normal de 41 semanas, nacida por cesárea a causa de sufrimiento fetal agudo (líquido meconial+++), peso al nacer de 3910 g, perímetro craneal de 37 cm y puntuación de Apgar 9-9.
A los 4 meses de edad se le detecta tortícolis izquierda, desviación de la mirada hacia arriba e hipotonía. Se realiza tomografía axial computadorizada (TAC) de cráneo y se observan signos discretos de atrofia cortical frontal. A los 7 meses se inicia en el programa de intervención integral temprana.
A los 15 meses se realiza resonancia magnética nuclear (RMN) de cráneo, que se informa como normal. Comienza la deambulación a los 18 meses y a los 22 meses de edad y los familiares notan que se detiene y se cae con mucha frecuencia, incluso en la posición sentada, se inclina hacia los lados por breves segundos, con frecuencia que fue aumentando hasta 20 veces en el día.
Fue valorada por su neurólogo y realizan electroencefalograma (EEG) de sueño a los 24 meses, que informa actividad paroxística de puntas aisladas en la región frontotemporal derecha y actividad de base normal. Se interpreta como epilepsia parcial compleja de corta duración y se comienza tratamiento antiepiléptico con los fármacos siguientes:
- Valproato de sodio (250 mg-5 mL) durante 1 mes; sin respuesta favorable.
- Valproato de magnesio (tableta de 190 mg) durante 1 mes; sin respuesta favorable.
- Depakine (solución, 200 mg/mL) durante 1 mes; sin respuesta favorable.
- Carbamazepina (tableta de 200 mg); se eliminó a los 9 días por empeoramiento de las crisis y signos de toxicidad.
A los 2 años y 4 meses se repite el EEG de sueño inducido, y se informa trastorno epileptiforme interictal muy activo en la convexidad parasagital, con predominio hacia las regiones frontales. Se comienza tratamiento con gabapentina, pero empeora el número diario de crisis, hay salivación abundante y pobre ganancia de habilidades motoras y de lenguaje.
Es evaluada en nuestro servicio a los 2 años y 5 meses de edad, y se realiza video-EEG, donde se constatan múltiples crisis electroclínicas de muy breve duración, con desviación de la mirada hacia arriba, pestañeo rítmico e inclinación lateral del tronco sin caídas, acompañadas de patrón eléctrico de punta-onda lenta de entre 3-4 ciclos por segundo (figura).
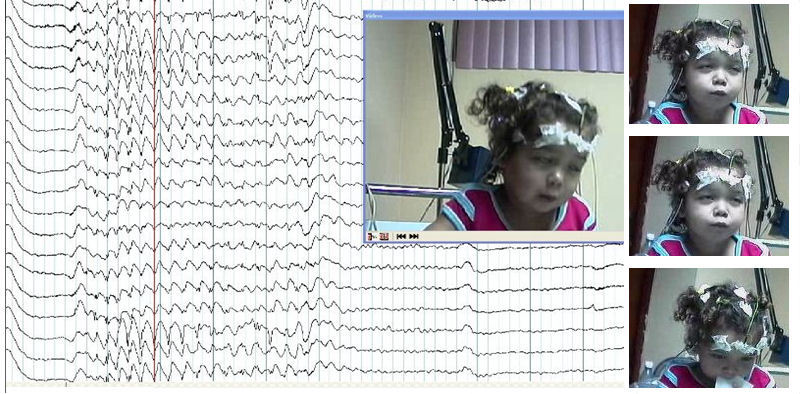
Figura. Trazado de videoelectroencefalograma durante la crisis electroclínica de ausencia. (La fotografía fue reproducida con el consentimiento informado del padre de la paciente).
Se retira rápidamente la gabapentina y se mantiene con depakine en solución. Se comienza tratamiento con lamotrigina a 0,1 mg/(kg · día), y se observa notable mejoría de las crisis, pero en la tercera semana de tratamiento reaparecen los episodios convulsivos con iguales características. Decidimos suspender lamotrigina y comenzar con solución de etosuximida, y se obtiene el control total de las crisis un mes después de iniciado el tratamiento. Se realiza vídeo-EEG evolutivo al mes del control total y resulta normal.
Actualmente se encuentra en monoterapia con etosuximida a 35 mg/(kg · día), con control total de las crisis y evidentes ganancias en el neurodesarrollo. Ha mejorado la tendencia a la desviación de la mirada hacia arriba.
DISCUSIÓN
Las epilepsias generalizadas idiopáticas (EGI) ocupan cerca de un tercio de las epilepsias infantiles13,14 y conforman un grupo heterogéneo de síndromes epilépticos posiblemente con patrones genéticos y mecanismos patogénicos diferentes que, aunque pueden darse a cualquier edad, tienen mayor expresividad en la edad escolar y la adolescencia. Las características diferenciales entre los distintos síndromes epilépticos son difíciles de establecer debido al solapamiento semiológico que puede darse entre ellos y, de hecho, el control evolutivo de estos pacientes suele inducir diferentes diagnósticos sucesivos que sugieren un continuum neurobiológico.10
En el niño, la epilepsia de ausencia infantil (EAI) es el único síndrome epiléptico, definido y admitido como idiopático en la clasificación internacional de epilepsias y síndromes epilépticos de 1989,15 que deja la puerta abierta a otros probables síndromes bajo la rúbrica de «otras epilepsias generalizadas no definidas anteriormente».16
Definir a nuestra paciente como portadora de una epilepsia idiopática es obviar elementos que desde el nacimiento nos orientan a la existencia de una causa subyacente. La presencia de tortícolis neonatal, hipotonía y retardo del neurodesarrollo, junto al inicio temprano de las crisis epilépticas, nos hacen pensar en una epilepsia generalizada probablemente sintomática.
En los últimos años se han descrito varios síndromes epilépticos considerados como EGI, al haberse observado que algunos pacientes epilépticos no podían encuadrarse correctamente en los síndromes existentes ya que presentaban un fenotipo clínico y electroencefalográfico de incierta valoración. Así, para algunos pacientes aparentemente afectos de una epilepsia de ausencia, en los que las crisis difieren por la duración o ciertos matices clínicos de las de la EAI típica, se ha debido buscar una nueva ubicación. El hecho de adscribirlos en la EGI se basa en criterios clínicos, electroencefalográficos, evolutivos y terapéuticos.16
El fundamento diagnóstico de la ausencia infantil es el registro electroencefalográfico, que gracias a la hiperventilación (HPV) mostrará las alteraciones críticas que, sobre una actividad de fondo normal, son los complejos de punta-onda a 3 Hz, que a veces se inician a 3,5 y finalizan a 2,5 Hz, síncronos bilaterales y con predominio frontal. El inicio y finalización son abruptos, y se recupera el registro sin depresión de voltaje ni enlentecimiento postictal.17,18 El inicio focal del trazado electroencefalográfico en nuestra paciente contribuyó al error diagnóstico de interpretarla como una epilepsia parcial compleja, dudas que pudieron solventarse gracias a la monitorización con vídeo-EEG, como ha sucedido a otros autores.6
La exacerbación de las crisis de ausencia por las drogas antiepilépticas ha sido demostrada con medicamentos como vigabatrina, carbamazepina y gabapentina.19 El empeoramiento de la epilepsia, inducido de manera paradójica por los propios antiepilépticos es una complicación clínica referida en los últimos años, en la que pueden estar implicados cuatro mecanismos patogénicos: intoxicación, elección inapropiada del fármaco, encefalopatía o efecto paradójico propiamente dicho.20
En las bases genéticas de la EAI se sugiere un locus principal en la región cromosómica 8q24 como rasgo de la epilepsia generalizada idiopática. Se estudian posibles loci en los cromosomas 5, 10, 13, 14 y 15, y alteraciones en los canales de diferentes iones o receptores de glutamato, entre otros.21-23
La epilepsia de ausencias, al estar genéticamente determinada, implica la ausencia de daño cerebral, aunque en algunas series se incluyen pacientes con retraso mental. En la actualidad, el pronóstico de la EAI se considera menos benigno que en otros tiempos. Cuando estos niños son adultos jóvenes, sobre todo aquellos en los que persisten las ausencias, tienen problemas psicosociales, particularmente en los estudios, en las relaciones sociales, de salud mental y en su comportamiento.24,25 La remisión de las crisis no asegura una buena evolución psicosocial, que puede plantear dificultades emocionales y psíquicas.9
Actualmente, el tratamiento que se elige para la EAI es el valproato, y la tendencia general es a utilizarlo en monoterapia, con lo que se controla a un 80 % de los pacientes.16,26 En los casos refractarios, se suele añadir etosuximida o lamotrigina. Está demostrada la buena tolerancia y la eficacia en monoterapia de la lamotrigina (LTG).26,27
La efectividad del tratamiento con etosuximida en nuestro caso nos confirma en primer lugar el diagnóstico sindrómico planteado, a pesar del inicio tan precoz de las manifestaciones convulsivas y confirma los recientes planteamientos en relación con la etiología poligénica de la epilepsia de ausencia y su estrecha vinculación etiológica con las canalopatías del sistema nervioso central.
Se ha señalado la importancia de los canales de calcio de tipo T en la sincronización de los circuitos tálamo-corticales y se ha propuesto que esta disfunción puede ser la responsable de los estados hipersincrónicos cerebrales, como los observados en la epilepsia de ausencia. Esta idea se sustenta por el descubrimiento de uno de los mecanismos de acción de drogas anti-ausencia, como la etosuximida, que es la supresión de los canales de calcio en las neuronas talámicas.28,29
El presente caso nos convoca a romper algunos esquemas diagnósticos, a la necesidad de evaluar evolutivamente a nuestros epilépticos de inicio y nos muestra una vez más, la necesidad de generalizar técnicas diagnósticas más precisas como el videolectroencefalograma.
summary
A case of absence epilepsy that began before the second year of life and required many antiepileptic drugs was presented. A literature review was made on this topic, delving into the present variations of diagnostic criteria related to epileptic absence syndromes and their various presentations that may derive from diagnostic and therapeutical mistakes. The important role of electroencephalographic and videoelectroencephalographic monitoring as a diagnosing tool in rare epilepsies in childhood was confirmed. The present etiological polymorphic factors, the role of ion channels and the use of antiepileptic drugs in infantile absence epilepsy were reviewed.
Key words: absence epilepsy, petit mal, etosuximide, calcium channels, videoelectroencephalogram.
REFERENCIAS BIBLIOGRÁFICAS
1. Mory S, Guerreiro CA, Li ML, Teixeira RA, Costa AL, Cardoso TA, et al. Epilepsias generalizadas idiopáticas diagnosticadas incorrectamente como epilepsias parciales. Arq Neuropsiquiatr 2002;60(3-B):788-796.
2. Panayiotopoulos CP, Tahan R, Obeid T. Juvenile myoclonic epilepsy: factors of error involved in the diagnosis and treatment. Epilepsia. 1991;32:672-676.
3. Panayiotopoulos CP, Obeid T, Waheed G. Differentiation of typical absence seizures in epileptic syndromes: a video EEG study of 224 seizures in 20 patients. Brain 1989;112:1039-1056.
4. Aliberti V, Grunewald RA, Panayiotopoulos CP, Chroni E. Focal electroencephalographic abnormalities in juvenile myoclonic epilepsy. Epilepsia. 1994;35:297-301.
5. Berkovic SF, Andermann F, Andermann E, Gloor P. Concepts of absence epilepsies: discrete syndromes or biological continuum. Neurology. 1987;37:993-1000.
6. Casas Fernandez C. Enfoque diagnóstico del niño con crisis generalizadas.Rev. Neurol 1998; 26: 311-21.
7. Cavazzuti GB, Ferrari P, Galli V. Epilepsy with typical absence seizures with onset during the first year of life. Epilepsia 1989; 30: 802-6.
8. Livingston S, Torres I, Pauli LL. Petit Mal epilepsy. Results of a prolonged follow-up study of 117 patients. J Am Med Assoc 1965; 194: 227-32.
9. Ureña-Hornos T, Rubio-Rubio R, Gros-Esteban D, Cabrerizo de Diago R, Peña-Segura JL, López-Pisón J. Epilepsia con ausencias. Revisión de nuestra experiencia de 14 años. Rev. Neurol 2004; 39: 1113-9.
10. Durá Travé T, Yoldi Petri ME. Ausencias típicas: características epidemiológicas, clínicas y evolutivas. An Pediatr (Barc)2006; 64(1):28-33.
11. Ferrie CD, Agathonikou A, Panayiotopoulos CP. Electroencephalography and video-electroencephalography in the classification of childhood epilepsy syndromes. J F Soc Med 1998;91:251-259.
12. Posner EB, Mohamed K, Marson AG. Ethosuximide, sodium valproate or lamotrigine for absence seizures in children and adolescents. The Cochrane Database of Systematic review 2005: issue 3.
13. Panayiotopoulos CP. Idiopathic generalised epilepsias. En: A clinical guide to epileptic syndromes and their treatment. Oxfordshire: Bladon Medical Publishing; 2002. p. 114-60.
14. Berg AT, Shinar S, Levy SR, Testa FM. Newly diagnosed epilepsy in children: Presentation at diagnosis. Epilepsia. 1999;40:445-52.
15. Commission on classification and terminology of the International League Against Epilepsy. Proposal for the revised classification of epilepsies and related syndromes. Epilepsia 1989; 30: 38998.
16. Nieto Barrera M. Síndromes epilépticos generalizados idiopáticos del niño. Rev. Neurol 2001;32: 650-9.
17. Aicardi J. Epilepsies with typical absence seizures. In Aicardi J, ed. 2 ed. Epilepsy in children. New York: Raven Press Books, Ltd.; 1994. p. 94-117.
18. Lockman LA. Ausencias. En Swaiman KF, eds. 2 ed. Neurología Pediátrica. Principios y prácticas. Madrid: Mosby/Doyma Libros; 1996. Pp.543-7.
19. Gelisse P, Genton P, Kuate C, Pesenti A, Baldy-Moulinier M, Crespel A. Seizure exacerbation by antiepileptic drugs.Epilepsia2004;45:1282–1286.
20. Rojo B, Arteaga R, Herranz JL. Empeoramiento de las epilepsias inducido por fármacos antiepilépticos. Bol Pediatr 1999; 39: 248-252.
21. Marini C, Harkin LA, Wallace RH, Mulley JC, Scheffer IE, Berkovic SF. Childhood absence epilepsy and febrile seizures: a family with a GABAA receptor mutation. Brain 2003; 126: 230-40.
22. Izzi C, Barbon A, Kretz R, Sander T, Barlati S. Sequencing of the GRIK1 gene in patients with juvenile absence epilepsy does not reveal mutations affecting receptor structure. Am J Med Genet 2002; 114: 354-9.
23. Celesia GG. Are the epilepsies disorders of ion channels? Lancet 2003;361: 1238-9.
24. Pavone P, Bianchini R, Trifiletti RR, Incorpora G, Pavone A, Pavano E. Neuropsychological assessment in children with absence epilepsy. Neurology 2001; 56: 1047-51
25. Olsson I, Campenhausen G. Social adjustment in young adults with absence epilepsies. Epilepsia 1993; 34: 846-51.
26. Campos-Castelló J, Prats-Viñas JM, García-Ribes A. Epilepsias idiopáticas: aspectos terapéuticos. Rev Neurol 2004; 38: 180-4.
27. Mauri-Llerda JA, Tejero-Juste C, Íñiguez C, Morales-Asín F. Utilidad de la lamotrigina en el tratamiento de las crisis epilépticas de ausencia. Rev Neurol 2001; 32: 247-50.
28. Huguenard JR. Block of T-type Ca2+ channels is an important action of succinimide antiabsence drugs. Epilepsy Currents 2002;2:49–52.
29. Vitko I, Chen Y, Arias JM, Shen Y, Wu XR, Perez-Reyes E. Functional Characterization and Neuronal Modeling of the Effects of Childhood Absence Epilepsy Variants of Cacna1h, a T-Type Calcium Channel. JNeurosci 2005;25(19):4844-4855.
Recibido: 8 de diciembre de 2006. Aprobado: 15 de febrero de 2007.
Dra. Ileana Valdivia Álvarez. Avenida 31 y 76, Marianao. La Habana, Cuba.
Correo electrónico: ileana.valdivia@infomed.sld.cu
1 Especialista de II Grado en Pediatría. Neuropediatra. Profesor Auxiliar de Pediatría.
2 Especialista de I Grado en Neurofisiología Clínica. Instructor de Neurofisiología.
3 Licenciado en Psicología. Asistente de la Universidad de La Habana.

