










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr v.80 n.2 Ciudad de la Habana abr.-jun. 2008
ORIGINAL
Características clínicas y evolución de la retinosis pigmentaria en los adolescentes
Clinical characteristics and evolution of retinosis pigmentosa in adolescents
Raisa Hernández Baguer,I Mirta Copello Noblet,II Bárbara Cid Vázquez,III Ana Maria Cabezas García,IV Daysi Domínguez RodríguezV y Beatriz Dyce GordonVI
I Especialista de II Grado en Oftalmología. Profesor Auxiliar. Centro de Referencia Nacional de Retinosis Pigmentaria. Hospital Docente "Dr. Salvador Allende". La Habana, Cuba.
IIEspecialista de II Grado en Oftalmología. Profesor Auxiliar. Centro de Referencia Nacional de Retinosis Pigmentaria. Hospital Docente "Dr. Salvador Allende". La Habana, Cuba.
IIILicenciada en Oftalmología y Optometría. Instructor. Centro de Referencia Pediátrico de Retinosis Pigmentaria. Hospital Pediátrico del Cerro. La Habana, Cuba.
IVEspecialista de I Grado en Oftalmología. Instructor. Centro de Referencia Pediátrico de Retinosis Pigmentaria. Hospital Pediátrico del Cerro. La Habana, Cuba.
VEspecialista de I Grado en Oftalmología. Centro de Referencia Pediátrico de Retinosis Pigmentaria. Hospital Pediátrico del Cerro. La Habana, Cuba.
VIEspecialista de I Grado en Genética Clínica. Centro de Referencia Nacional de Retinosis Pigmentaria. Hospital Docente "Dr. Salvador Allende". La Habana, Cuba.
RESUMEN
INTRODUCCIÓN. La retinosis pigmentaria es la causa más frecuente de distrofia retiniana. En la adolescencia comienzan enfermedades oculares de tipo distrófico, origen genético y curso progresivo. El objetivo del presente estudio fue identificar las características clínicas y evolución de la retinosis pigmentaria en la adolescencia.
MÉTODOS. Se realizó un estudio descriptivo y prospectivo en jóvenes de edad entre 10 y 14 años (Grupo I), y entre 15 y 19 años (Grupo II), de uno y otro sexo, que fueron atendidos en el Centro de Retinosis Pediátrico Nacional entre 2002 y 2006. Se observó herencia, síntomas, edad, tiempo transcurrido, enfermedades generales y electrorretinograma. Los medios transparentes, fondo de ojo, biomicroscopia y perimetría se hicieron al inicio, al año y al final del estudio. Se clasificaron los casos en 4 estadios atendiendo a la agudeza y campo visual.
RESULTADOS. Predominaron los pacientes del sexo masculino y color blanco de la piel. Se hallaron opacidades del cristalino en 61 ojos (71,4 %), principalmente en el grupo 2. Al final del estudio 32 adolescentes (76,2 %) no tuvieron evolución progresiva; 5 (11,9 %), medianamente progresiva y 5 (11,9 %), muy progresiva, con marcado empeoramiento de la función visual.
CONCLUSIONES. Las características de la retinosis pigmentaria identificadas en la mayoría de los jóvenes concuerdan con los primeros estadios de la enfermedad. La evolución durante 5 años no ha sido progresiva en la mayoría de los adolescentes de uno y otro grupo, lo que nos hace considerar que los cambios biológicos de esta etapa de la vida, aparentemente no influyen empeorando el curso de la enfermedad ocular.
Palabras clave: Retinosis pigmentaria, adolescencia, ceguera nocturna, distrofia retiniana.
ABSTRACT
INTRODUCTION. Retinosis pigmentosa is the most frequent cause of retina dystrophy. Ocular diseases of dystrophic type, genetic origin and progressive course appear in adolescence. The objective of the present study is to identify the clinical characteristics and evolution of retinosis pigmentosa in adolescence.
METHOD. A descriptive and prospective study was conducted among young subjects aged 10-14 (group 1) and 15-19 (group II) of both sexes that were seen at the National Pediatric Centre of Retinosis between 2002 and 2006. Heredity, symptoms, age, time elapsed; general diseases and electroretinogram were observed. The transparent means, fundus oculi, biomicroscopy and perimetry were performed at the beginning, at a year and at the end of the study. Taking into consideration the acuity and visual field, the cases were classified into 4 stages.
RESULTS. White males prevailed. Opacities of the crystalline were found in 61 eyes (71.4 %), mainly in group 2. At the end of the study, 32 adolescents (76.2 %) had no progressive evolution; 5 (11.9 %), fairly progressive; and 5 (11.9 %), very progressive, with a marked worsening of the visual function.
CONCLUSIONS. The characteristics of retinosis pigementosa identified in most of the youth agreed with the first stages of the disease. The evolution has not been progressive in most of the adolescents of both groups, which makes us think that the biological changes of this stage of life do not apparently worsen the course of the ocular disease.
Key words: Retinosis pigmentosa, adolescence, night blindness, retinal dystrophy.
INTRODUCCIÓN
La adolescencia es un período de la vida del ser humano caracterizado por rápidos y diversos cambios que se suceden de forma muy dinámica, cambios físicos y emocionales fundamentales que crean las características de la vida adulta ulterior.1
Una edad tan compleja y trascendental requiere también una atención consecuente. De un modo convencional se considera que la adolescencia es el período que cubre la segunda década de la vida: de los 10 a los 20 años. No existe otro momento después del nacimiento en que los hechos biológicos se produzcan tan rápido.2
En esta segunda década de vida tienen su inicio enfermedades oculares de tipo distrófico, genéticamente determinadas y cuya naturaleza familiar ha sido conocida por más de 100 años. Entre estas enfermedades oculares distróficas de origen genético y evolución crónica se encuentra, en primer lugar, la retinosis pigmentaria (RP), que agrupa un conjunto de enfermedades que de forma primaria y progresiva afectan a los fotorreceptores (conos y bastones) y después otras capas de la retina que ocasionan ceguera nocturna, disminución de la visión periférica y posteriormente de la visión central, con lo que da lugar a una discapacidad visual con características muy particulares.3
Otra característica clínica es la posible concomitancia de la RP con enfermedades endocrinas, trastornos auditivos, neurológicos, entre otros, que conforman verdaderos síndromes. Se trata de la RP sindrómica o asociada.3
En su curso natural, la RP puede comenzar a cualquier edad y llegar hasta la ceguera total en un tiempo variable, lo cual en cierta medida depende de la edad de comienzo y de la gravedad del tipo de RP. Sin embargo, la literatura médica mundial muy poco se ha referido a las características de la enfermedad durante la etapa de la adolescencia.4
La prevalencia mundial de la RP se reconoce de 1 por 5000 habitantes y se considera que una de cada 80 personas presenta el gen causante de la enfermedad. En Cuba, por las particularidades del Sistema Nacional de Salud y por la labor de pesquisa desplegada en la población cubana por la Red Nacional del Programa de Atención a pacientes con RP y sus familiares, se conoce que la prevalencia es de 4,8 por 10 000. Debido a las limitaciones que la RP ocasiona al individuo que la padece y que lo puede llevar a la discapacidad o a la minusvalía, es necesario reconocer que ella repercute indiscutiblemente en la actitud, estilo y calidad de vida del afectado. La actitud ante la enfermedad no es igual si se trata de un niño, un adolescente o un adulto; así también es muy personal la comprensión, aceptación y adaptación a la enfermedad.5,6
La RP en Cuba se ha insertado prioritariamente en el marco de la pesquisa activa de enfermedades visuales en mayores de 5 años de edad y en el programa de salud para discapacitados, del cual no hay similar en el mundo. El objetivo de este estudio fue identificar las características y la forma de evolución clínica de los adolescentes con RP, lo cual permitió ganar en información y comprensión acerca del comportamiento de este padecimiento en esta etapa de la vida.
MÉTODOS
Se realizó un estudio descriptivo, longitudinal y prospectivo en adolescentes con RP entre 10 a 19 años (según criterio de la OMS) que se atendieron en el Centro de Referencia Nacional y Pediátrico de Retinosis Pigmentaria desde el 2002 hasta el 2006, con edad límite de captación para el estudio de 15 años, ya que se requería que aún fueran adolescentes una vez transcurridos los 5 años de la exploración. Este fue el criterio se de inclusión en el estudio. Se excluyeron los casos que no cooperaron bien para los exámenes o cuyos familiares no quisieron que continuaran en el estudio.
Se recogió en encuesta individual los datos que permitieron determinar las características clínicas de la enfermedad en cada adolescente, la conclusión de los exámenes oftalmológicos al inicio, anualmente y al final del estudio para una vez procesados, ofrecer los resultados correspondientes según los objetivos de la investigación. Para determinar los antecedentes clínicos y las características de la RP se tuvieron en cuenta: tipo de herencia, síntoma inicial y edad de inicio, tiempo de evolución, enfermedades generales y oculares concomitantes y las particularidades de los medios transparentes y de las estructuras del fondo de ojo detallados por biomicroscopia y oftalmoscopia directa, y en algunos casos, indirecta, si lo permitía la cooperación del paciente.
Estos signos fondoscópicos definen la forma clínica de RP de que se trata, por lo que fueron explorados y evaluados por ojo examinado, puesto que se trata de establecer la progresión de la enfermedad ocular. Se confirmó el diagnóstico por electrorretinografía con equipo computadorizado EREV 2000 Plus de la LACE y se realizó perimetría cinética tipo Goldmann con estímulo blanco de 64 mm² y filtro de 1,00 con Suma V/4 preferentemente, para determinar los límites de la visión periférica, o sea el campo visual.
Se clasificó el estadio clínico en 4 etapas atendiendo a la agudeza visual con corrección y al área de campo visual residual que presentaba el mejor ojo de cada adolescente; se consideró como elementos colaterales el estado de los medios transparentes y los signos de la RP en el fondo de ojo. La clasificación responde a la metodología de trabajo de la Escuela Cubana de RP fundada por el Dr.C. Prof. Orfilio Peláez Molina (1923-2001) en 1989 y utilizada para evaluar estos parámetros principales y calificar el estadio y dar pronósticos evolutivos. Los estadios clínicos son:7
I con AV ³ 0,5 y CV ³ 15°
II con AV ³ 0,3 y CV entre 11° - 15°
III con AV ³ 0,05 y CV entre 5° - 10°
IV con AV £ 0,05 y CV - de 5° considerando el ojo mejor
Para identificar la forma de evolución de la RP se tomaron en cuenta la variación de los parámetros principales interexamen anual y los cambios en el estadio clínico de la enfermedad; se comparó los resultados obtenidos al inicio y al final del estudio en cada adolescente. Cada tipo de RP (típica, atípica o asociada) y de herencia (autosómica dominante, autosómica recesiva, o recesiva ligada al cromosoma X= AD, AR y RLX respectivamente) fueron bien definidas.
Para la clasificación de los síndromes asociados se tuvo en cuenta para el Usher la clasificación de Kimberling donde se estableció que el Usher I es aquel donde concomitan sordera congénita y retinosis pigmentaria, y el Usher II cuando la hipoacusia es moderada acompañada de la RP.8,9
Se establecieron tres formas de evolución:
1. No progresiva: no hubo cambio de estadio clínico.
2. Medianamente progresiva: hubo cambio al estadio clínico siguiente de la clasificación.
3. Muy progresiva: hubo cambios a estadios más avanzados de la clasificación.
Esta calificación permitió conocer el comportamiento de la RP en la etapa de la adolescencia en cada uno de los jóvenes y una vez transcurridos los 5 años del estudio, posibilitó conocer la posible influencia de los cambios biológicos y psíquicos de esta etapa sobre el curso de la enfermedad ocular.
Se obtuvo la distribución de acuerdo con cada variable definida; se utilizó como medida de resumen el porcentaje y se expuso los resultados en tablas y gráficos.
Operacionalización de las variables:
| Variables | Tipo | Escala | Descripción | Indicadores |
| Edad | Cuantitativa continua | 10-14 | Adolescentes según edad biológica y criterio de la OMS | - % por grupo de edades |
| Color | Cualitativa nominal | Blanca | Según color | Porcentaje según |
| Sexo | Cualitativa Nominal | Femenino | Según sexo biológico | Porcentaje según |
| Inicio | Cualitativa Nominal Politómica | - Precoz | Referido por la H.C. | Porcentaje según |
| Tipo de RP según la clínica | Cualitativa Nominal |
| Referido por la H.C. | Porcentaje según |
| Estadio clínico | Cualitativa Ordinal | I | Según clasificación de la Escuela Cubana de Retinosis Pigmentaria | Porcentaje según |
| Tipo de herencia presente | Cualitativa Nominal Politómica | Autosómica Dominante. | Según el patrón que se presente en cada paciente | Porcentaje según |
| Evolución | Cualitativa Nominal | No progresiva | Según cambios de la AV-CV y de estadio clínico | Porcentaje según |
*Monocular, en sector, central o inversa, paravenosa o sin pigmentos.
**Acompañando a otras enfermedades como obesidad, sordera parcial o total, enfermedades neurológicas, trastornos psiquiátricos, etc.
RESULTADOS
La muestra fueron 42 adolescentes de uno y otro sexo, que en el estudio se distribuyeron en dos grupos: I y II. Predominaron los varones en ambos grupos y se tuvo el criterio de que:
- El Grupo I con edades entre 10-14 años de edad, corresponde a la adolescencia temprana, con promedio de edad de 12,3 (DS 2,2)
- El Grupo II con 15 a 19 años de edad, corresponde a la adolescencia media hasta la tardía.2 El promedio de edad es de 16,8 (DS 1,2) (figura 1).

Predominó en los adolescentes la piel blanca para un 61,9 % y solo el 9,6 % tuvo la piel de color negro.
Atendiendo a los antecedentes clínicos de la RP, la edad en que aparece el primer síntoma, es decir la ceguera nocturna, se señala como el inicio de la enfermedad, que fue precoz antes de los 10 años de edad en 30,9 % y juvenil después de los 10 años en 69,1 %, por lo cual los adolescentes estudiados tenían un tiempo de evolución de la RP entre 5 y 10 años (DS 2 ,1) ya al inicio del estudio.7
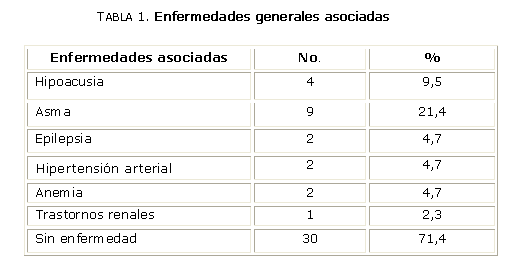
Los adolescentes presentaron diferentes enfermedades generales antes del inicio del estudio, lo que se informó al interrogarlos sobre los antecedentes patológicos personales (APP) y otras afecciones ocurrieron en el transcurso de la investigación. La tabla 1 refleja solamente las enfermedades de interés, porque pueden formar parte junto con la RP de síndromes asociados. Las entidades RP e hipoacusia neurosensorial ocasionan la doble discapacidad de sordoceguera y apuntan hacia la presencia de síndrome de Usher en 3 pacientes, pues 1 presentaba hipoacusia por ototoxicidad medicamentosa.9

El hábito de fumar estuvo presente en 7 adolescentes varones (16,6 %) que formaban parte del grupo II.
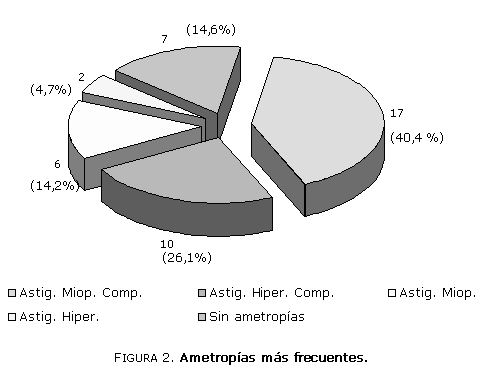
En el 71,4 % de los jovenzuelos existían ametropías (figura 2) y en 10 de ellos (23,8 %) estrabismos latentes y del tipo acomodativo, que no se corrigen con tratamiento quirúrgico. Las ametropías son la causa más frecuente por las que los niños y jóvenes acuden a las consultas de oftalmología. Por ello es importante reconocer qué tipo de defecto predomina entre ellos y debe ser siempre corregida cuanto antes con el método que se prefiera.

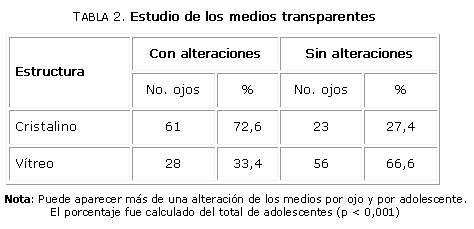
Las alteraciones del vítreo y del cristalino no se observaron en jóvenes sanos (10), por lo cual se confirmó que la temprana aparición de estas afecciones en los adolescentes estuvo relacionada con diversos factores en el transcurso de la RP (tabla 2) (p < 0,001).
El análisis se hizo en 84 ojos. El desprendimiento de vítreo posterior estuvo presente en 12 ojos (14,2 %), los cambios en la transparencia del cristalino en forma de vacuolas y la catarata pequeña polar posterior (10) se detectaron en 61 ojos (72,6 %) al final del estudio, principalmente en adolescentes del grupo II.
En cuanto a la distribución de los 42 pacientes según las formas clínicas de la RP, se halló que la forma clínica típica estaba en 36 de ellos para el 85,7 %, fue la predominante por la presencia y la disposición de los pigmentos en el fondo de ojo de los pacientes. Las RP atípicas (7,1 %) fueron del prototipo apigmentos en 2 pacientes e inversa en 1 paciente. La RP asociada (Usher de tipo I y II) que fue hallada solamente en 3 enfermos; clínicamente mostró características típicas al fondo de ojo también (7,1 %).
En relación con el tipo de herencia, destacó la AD con 11 adolescentes (26,1 %), la AR con 21,4 % al igual que la RLX porque en ambas se ubicaron a 9 jóvenes. En 13 adolescentes la herencia no pudo ser definida para un 30,9 %.
En 10 adolescentes (11,9 %) hubo cambios al final del estudio en la agudeza y campo visuales, quedó 5 de ellos dentro del grupo de débiles visuales según la clasificación de la OMS, con 0,3 con corrección o campo visual reducido entre 5 y 10 grados en el mejor ojo, lo cual implica una pérdida visual de un 25 % a 30 % para la lectura y de 84 % de campo visual aproximadamente.11
Desde el punto de vista de la evolución por formas clínicas esta fue "no progresiva" en casi todos los pacientes con RP típica, en las atípicas fue "progresiva" en 2 de 3 jóvenes. La evolución de la RP asociada al final del estudio fue "moderadamente progresiva" solo en 1 adolescente.
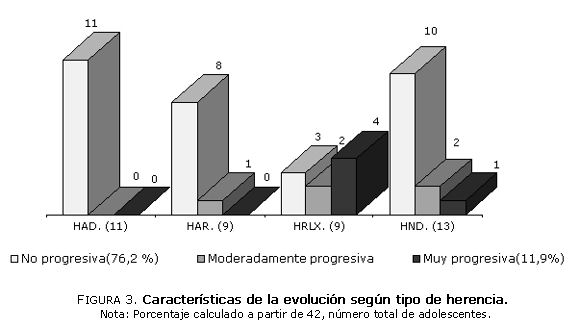
En la figura 3 se muestra la evolución clínica de la enfermedad según el tipo de herencia, en 6 de los 9 pacientes con herencia recesiva ligada al X hubo progresión moderada en 2 casos y francamente tórpida en 4 casos. En 1 caso en que la evolución fue también progresiva, el tipo de la herencia no pudo ser definida.
Al final del estudio en relación con la función visual según agudeza y campo visual como criterios mayores, 32 adolescentes (76,2 %) se mantuvieron estables o con evolución no progresiva, en 5 casos (11,9 %) la evolución fue medianamente progresiva y con la misma cifra hubo marcado empeoramiento de esta función y por tanto la evolución de la RP fue muy progresiva.
DISCUSIÓN
Los 42 adolescentes que se atendieron en los Centros de Referencia Pediátrico y Nacional, una vez confirmado el diagnóstico de RP han recibido el tratamiento cubano para esta enfermedad, terapéutica que comprende ozonoterapia, electroestimulación, vitaminoterapia y cirugía revitalizadora.12,14 Este procedimiento corresponde a la metodología de trabajo de los Centros de la Red y el Programa Nacional de Retinosis Pigmentaria en Cuba, que no fueron dejados de realizar al llevar adelante esta investigación contando con el consentimiento informado de pacientes y familiares.
Se consideró necesario hacer esta aclaración, ya que evaluar el resultado del tratamiento médico aplicado, no fue objetivo de esta investigación.
Aunque los cambios propios de la adolescencia se van presentando paulatinamente durante los años que enmarcan esta etapa, se decidió agruparlos por edades similares atendiendo a lo que se llama adolescencia temprana y tardía. 2,15
En relación con el color de la piel, se reportó que la RP afecta de forma mayoritaria a los individuos de color blanco y al género masculino. Esto último por existir la herencia recesiva ligada al X donde los enfermos son hombres solamente incrementó la población de enfermos de este género. 16
En cuanto al color de la piel, las investigaciones y bibliografías revisadas se refieren de forma prevalente a sitios del mundo donde las etnias de color de piel blanca son las imperantes. Quizás si estas investigaciones y publicaciones se hicieran o lleguen procedentes de otros países, donde las poblaciones tengan otro color de la piel se pueda conocer si prevalecen en ellos los enfermos de RP.
Al precisar la edad de inicio de la enfermedad, al comienzo del estudio, se advirtió que algunos pacientes habían aplazado la asistencia inmediata a la consulta de Oftalmología una vez aparecidos los síntomas a pesar de que el porcentaje de población atendida por médicos de la familia fue de 99,7 % como señalan los indicadores básicos del MINSAP para el 2006. 17 Otros fueron diagnosticados por la pesquisa activa que se realizó desde años atrás o por el estudio de familias afectadas ya conocidas. Este proceder contempló la prevención y manejo de la enfermedad que divulgan la Escuela y el Programa para la atención de la Retinosis Pigmentaria en Cuba.
En el examen de los medios transparentes, la córnea no presentó alteraciones, en cambio las modificaciones vítreas se descubrieron más tempranamente que las del cristalino. Se observó celularidad, desorganización fibrilar y flóculos en ese orden de aparición. Es prácticamente imposible observar este resultado si en la RP no se afectara verdaderamente los medios transparentes. Estudios epidemiológicos (Harding, 1991) señalaron el aumento en la incidencia de las opacidades del cristalino en las distintas distrofias vítreo retinianas y que está presente en la mayor parte de los afectados.18
La presencia de pigmentos retinianos, los cambios en el epitelio pigmentario dio un aspecto no homogéneo al fondo de ojo como granulación fina y el estrechamiento del calibre vascular son los signos más precoces del RP en los adolescentes con menos tiempo de evolución de la enfermedad.19,20
De forma más tardía aparecieron las variaciones del color del disco óptico y del aspecto de la mácula entre 7 u 8 años de evolución de la enfermedad. Todos estos cambios pueden verse de forma bilateral y bastante simétrica en el fondo de ojo.21
Se hallaron otras enfermedades oculares que acompañó a la RP como las ametropías y dentro de estas el astigmatismo miópico compuesto, que fue el más frecuente. El estrabismo latente que tiene incidencia mayor en la etapa preescolar y escolar, también se manifestó. El nistagmo tuvo relación con el tipo de RP central o inversa, por mala fijación por el daño macular se presentó en 1 adolescente.22
Solo el síndrome de Usher en sus formas clínicas I y II, según la clasificación de Kimberling, estuvo presente como RP sindrómica o asociada. Esta forma es la más frecuente también a nivel mundial.8
El determinar si la RP es parte de un síndrome o no será consecuencia del estudio y la observación del paciente durante años. Otros padecimientos como afecciones ortopédicas, estomatológicas, psicológicas, infectocontagiosas, dermatopatías, ginecológicas, entre otras; propias de esta etapa de la vida y que forman parte de los índices de morbilidad, fueron debidamente recogidas en encuestas individuales y atendidos como está orientado en el Programa Nacional de Atención Integral a la Salud de Adolescentes. MINSAP, Cuba 2000.23,26
En 2 casos se presentó de formas clínicas atípicas y en 1 asociado en los que hubo mayor y moderada progresión de la enfermedad respectivamente, esto fue quizás porque los cambios importantes sean por una toma temprana de la retina central macular como se reporta en las RP atípicas o por los cambios neurosensoriales en las asociadas. Se dañan además la visión de colores y la lectura cercana, lo cual repercute más en la vida y desempeño del paciente que si fuera la forma típica donde el daño es en la visión periférica y por tanto en el extremo campo visual.
Finalmente, se quiso precisar si existía alguna relación entre la forma de evolución de la RP y el tipo de herencia, se observó que los adolescentes con herencia autosómica dominante revelaron durante el tiempo del estudio una demostrada estabilidad de la enfermedad al no encontrarse cambios con relación al estadio clínico inicialmente determinado.
Algunos autores describen la RP de herencia autosómica dominante como forma benigna de la enfermedad, puesto que en edades avanzadas y aún después de operarse de catarata, en el caso de los adultos, se conserva de moderada a buena agudeza visual central y campo visual aceptable para la orientación y desplazamiento. En el estudio el pronóstico visual para este tipo de herencia fue favorable.27
El tipo de RP ligado al X se describió como la forma más severa de la enfermedad degenerativa retiniana que conllevó a marcada disminución de la agudeza visual en edades tempranas.16,18 los adolescentes con este tipo de herencia se encontraron ubicados en ambos grupos de edades y presentaron en su mayoría los estadios clínicos II y III de la RP a pesar de su corta edad.
Hubo progresión de la RP también en 3 de los 13 adolescentes que no tenían precisado el tipo de herencia, puede tratarse en un futuro de los tipos autosómico recesivo o recesivo ligado al cromosoma X, lo cual no pudo ser determinado mediante el estudio del árbol genealógico en ese momento.29,30
En el estudio realizado por Berson durante 3 años de observación del curso natural de la RP, en 24 pacientes de 6 a 17 años encontró la pérdida del área del campo visual en un 4,6 % por año los casos estudiados, lo que marcó una progresión de la enfermedad mucho mayor que la observada en nuestros enfermos.31
El resultado de las tonometrías arrojó una presión intraocular (PIO) dentro de límites normales en aquellos jóvenes en que su cooperación permitió la realización de este examen. Esto permitió descartar la hipertensión ocular piedra angular del glaucoma, como posible causa del empeoramiento de la visión en los casos en que así sucedió. Recordemos la ya descrita asociación de glaucoma en el 3 % de los afectados de RP.31
Se concluye que las características de la RP observadas en los adolescentes durante los 5 años del estudio, se identificaron con las enmarcadas en los primeros estadios de nuestra clasificación evolutiva de esta enfermedad ocular a pesar del tiempo de inicio y transcurso de la afección.
En la mayoría de los adolescentes la forma clínica típica fue la que prevaleció, tanto en los casos característicos como en los asociados, se considera que la forma apigmentos va relacionada más con el corto tiempo de evolución de la enfermedad transcurrido que con una atipicidad verdadera.
La evolución de la enfermedad ocular no ha sido progresiva en la mayoría de los adolescentes de ambos grupos, lo que hace considerar que los importantes cambios biológicos que forman parte de la adolescencia al parecer no influyen sobre el curso de la RP en esta etapa de la vida.
La relación entre formas de evolución y tipo de herencia fue significativa en los adolescentes varones. En los de herencia recesiva ligada al cromosoma X hubo empeoramiento de la agudeza y campo visuales sin existir otra causa ocular o extraocular que explique este acontecer. Esto sugiere que existe una evolución de la enfermedad muy particular para este tipo de herencia.
La recomendación para mejorar este trabajo en un futuro es mantener estrechos lazos con la Atención Primaria de Salud para efectuar la captación anticipada de los adolescentes con RP dentro de las familias afectadas, al analizar el tipo de herencia y determinar tempranamente quienes son las personas con factores de riesgo para hacer un diagnóstico precoz de la enfermedad. Así también, no se debe olvidar de la posible aparición en la infancia y la adolescencia de manifestaciones de RP sindrómica y que la más frecuente en el medio se relaciona con los sordos e hipoacúsicos.
REFERENCIAS BIBLIOGRÁFICAS
1. Canessa, Nykiel. Manual para la educación en salud del adolescente. OPS/UNFAP; 2004.
2. Valdés Martín S, Gómez Vasallo A. Temas de Pediatría. La Habana: Editorial Ciencias Médicas;2006.
3. Gutiérrez Torres SM. Retinosis Pigmentaria. Clasificación y tratamiento. 2004 [serie en Internet]. [Citado 12 julio 2006] Disponible en: http://retinois.org/olo/libro/l.htm
4. Hernández Baguer R, Copello M, Rubio L. Retinosis pigmentaria y adolescencia. Avances Médicos de Cuba. 1999;VI(17):58-61.
5. Sit AJ, Chipman M, Trope GE. Blindness registrations and socioeconomic factors in Canada: an ecologic study. Ophthalmic Epidemiology. 2005;12. [serie en Internet]. [Citado 3 de agosto 2007] Disponible en: http://www.tandf.co.uk/jounals/titles/09286586.asp
6. MINSAP. Cuba. Programa Nacional de Retinosis Pigmentaria. Reporte Nacional. MINSAP; 2007.
7. Herrera Mora M. Clasificación. En: Peláez Molina O. Editors Retinosis Pigmentaria. Experiencia Cubana. La Habana: Científico Técnica;1997. p.68-81.
8. Kimberling W. Smith RJH. Gene Mapping of the Usher Syndromes. Otolaryngology Clinic of North America. 2003;25(5):20-3.
9. Melamud A, Wang Q, Traboulsi EI. Retinitis pigmentosa and allied disorders. In: Huang D, Kaisr PK, Lowder CY, Traboulsi EI, editors. Retinal Imaging. Philadelphia: Mosby Elsevier; 2006. p.461-7.
10. Weleber RG, Gregory-Evans K. Retinitis pigmentosa and allied disorders In: Ryan's editors Vol I 4th. Ed Philadelphia: Elsevier Inc;2006. p.299-320.
11. Estándares visuales. En: Vaughan D, Asbury T, Riordan-Eva P. Oftalmología general. México, DF: El Manual Moderno; 2004. p.487-90.
12. Santiesteban Freixas R. Afecciones de la retina. En: Colectivo de autores editors Pediatría Tomo I. La Habana: Editorial Ciencias Médicas;2006.
13. Peláez O. Tratamiento de la RP. En: Peláez Molina O editors Retinosis Pigmentaria. Experiencia Cubana. La Habana: Científico Técnica;1997. p.173-8.
14. Travieso Y, Posada A, Fariñas L, Meléndez M, Martínez Z, Dujarric M. Empleo de las sustancias neurotróficas como terapéutica en la retinosis pigmentaria. Rev Cubana Invest Biomed. 2005;24(2):68-75.
15. Consuegra R. Problemas médicos de los adolescentes. La Habana: Científico Técnica;1999.
16. Kaplan J, Borneau Q. Clinical and Genet Heterogeneity in Retinitis Pigmentosa. Hum. Genet 1998;85:635-42.
17. Situación de salud en Cuba. Indicadores Básicos del MINSAP. MINSAP; 2006.
18. Kanski J.J. Oftalmología Clínica. Barcelona: Doyma;2004.
19. Traboulsi EI. A compendium of inherited disorders and the eye. New York: Oxford University Press;2006. p.234.
20. Hollyfield JG, Anderson RE, LaVail MM. Retinal degenerative diseases. New York: Springer;2006. Pp.557-559.
21. Gil-Gibernau J. El fondo de ojo en el niño. La Habana: Científico Técnica;1999.
22. Saw Seang-Mei, Chan Shih-Yen E, Koh A, Tan D. Interventions to retard myopia progression in children: an evidence-based update. Ophthalmology 2002;109(3):415-8.
23. Behrman RE, Kliegman RM, Jonson HB. editors Nelson, Tratado de Pediatría. Madrid: McGraw Hill; 2000.
24. Programa de desarrollo 2000: Pediatría/MINSAP. Cuba. Ciudad de La Habana: Ciencias Médicas; 2000.
25. Alvarez Sintes R. Atención Integral de Salud. Atención al adolescente En: Temas de Medicina General Integral. La Habana: Editorial Ciencias Médicas; 2001. p.159-64.
26. Roselot, J. Adolescencia. Problemática de salud del adolescente y joven en Latinoamérica y el Caribe. Pediatría. 2da Ed.Menchelo: Intermédica; 1999. Pp.25-7.
27. Rimoin DL, Connor JM, Pyeritz RE, Korf BR. Emery's and Rimoin´s. Principles and Practice of Medical Genetics. 4 Th Ed. Edinburgh London: Churchill Livingstone; 2002.
28. Melamud A, Shen GQ, Chung D, Si Q, Simpson E, Li L, Peachey NS, Zegarrra H, et al. Mapping a new genetic locus for X linked Retinitis Pigmentosa to Xq28. Journal of Medical Genetics. 2006 Jun;43(6):27-9.
29. Sack George H. Genética Médica. México, DF: Mc Graw Hill Interamericana;2002.
30. La Retinosis Pigmentaria en España: Estudio Clínico y Genético. Ed. Organización Nacional de Ciegos Españoles (ONCE). 2007 [serie en Internet]. [citado el 4 de Octubre 2007] Disponible en: http://www.retinosis.org
31. Berson E, Sandberg MA, Rosner B, Buch DB, Hanson A. Natural course of RP over a three year interval. Am J Ophthalmol. 1985;99:240-3.
32. Peng T, Wul, Zhou W. Retinitis Pigmentosa Associated with glaucoma. Clinical analysis Eye Sci. 1998;6(1):217-9.
Recibido: 10 de enero de 2007.
Aprobado: 26 de junio de 2007.
Dra. Raisa Hernández Baguer. Calzada del Cerro Núm. 1551. Ciudad de La Habana, Cuba. Correo electrónico: raisa.baguer@infomed.sld.cu


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}