










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr v.80 n.4 Ciudad de la Habana oct.-dic. 2008
Síndrome de Aarskog: hallazgos fenotípicos en una cohorte de pacientes
Aarskog syndrome: phenotypic findings in a cohort of patients
Iván Hernández García,I Débora A. García Martínez,II Mayra Hernández Iglesias,III Norma Elena de León Ojeda,IV Lourdes Fátima Marrón Portales V
I Especialista de I Grado en Genética Clínica. Instructor. Servicio de Genética, Hospital Pediátrico Docente «William Soler». La Habana, Cuba.
II Especialista de II Grado en Neonatología. Profesora Titular. Máster en Atención Integral al Niño. Servicio de Genética, Hospital Pediátrico Docente «William Soler». La Habana, Cuba.
III Licenciada en Biología. Máster en Antropología Biológica. Asistente. Servicio de Genética, Hospital Pediátrico Docente «William Soler». La Habana, Cuba.
IV Especialista de II Grado en Genética Clínica. Asistente. Servicio de Genética, Hospital Pediátrico Docente «William Soler». La Habana, Cuba.
V Licenciada en Tecnología de la Salud. Instructora. Servicio de Genética, Hospital Pediátrico Docente «William Soler». La Habana, Cuba.
RESUMEN
Se presentan los hallazgos fenotípicos de 7 pacientes diagnosticados de síndrome de Aarskog en el servicio de Genética del Hospital Pediátrico Docente «William Soler». Las características fenotípicas que estuvieron presentes en todos los pacientes fueron el pico de viuda, las extremidades cortas y la braquidactilia. Les siguieron en orden de frecuencia (85 %) la nariz pequeña, el puente nasal ancho y el filtrum largo y ancho. Otras dismorfias faciales encontradas (71 %) fueron la frente amplia, las narinas antevertidas, el hipertelorismo, el cuello corto y el surco simiano. Se discute la posibilidad de que exista algún sesgo de detección debido a que el examinador presta mayor atención a la cara que a otros segmentos corporales. La criptorquidia, que distingue y da nombre al síndrome, se encontró en un porcentaje menor de individuos (60 %). En dos casos se encontró hipoplasia renal, hallazgo ocasional en la literatura consultada. En ningún caso se constató retraso mental. Las diferencias fenotípicas halladas pudieran atribuirse a las diferencias moleculares reportadas en la literatura.
Palabras clave: Síndrome de Aarskog, baja talla, criptorquidia, displasia fascio-digito-genital.
ABSTRACT
The phenotypic fndings of 7 patients who were diagnosed Aarskog syndrome in the Service of Genetics of «William Soler» Pediatric Teaching Hospital were presented. The phenotypic characteristics appearing in all patients were widow's peak, short extremities and brachydactilia. They were followed in order of frequency (85 %) by small nose, wide nasal bridge and long and wide filtrum. Other facial dismorphies (71 %) were wide forehead, anteverted narines, hypertelorism, short neck and simian crease. It is discussed the possibility that there is some bias of detection due to the fact that the examiner pays more attention to the face than to other body segments. Criptorchydia, that distinguishes and gives name to the syndrome, was found in a lower percent of individuals (60 %). Renal hypoplasia, an occasional finding in the consulted literature, was observed in 2 cases. Mental retardation was not confirmed in any case. The phenoptypic differences detected could be attributed to mollecular differences reported in literature.
Key words: Aarskog syndrome, low height, cryptorchidia, faciodigitogenital dysplasia.
INTRODUCCIÓN
El síndrome de Aarskog, también conocido como displasia fascio-digito-genital es una entidad monogénica, de herencia dominante ligada al X, causada por mutaciones en el gen FGD1 (displasia fascio genital-1) situado en el locus Xp11.21.1 El producto del FGD1 contiene los dominios de homología Dbl (DH) y plekcistrina (PH).2 Este transcripto se une específicamente a una proteína Rho, familia de GTPasa llamada Cdc42Hs y estimula el intercambio GDP-GTP de las formas isopreniladas de la Cdc42Hs y de igual modo aumenta la actividad mitogénica en la cascada de las proteínas quinasas, entre estas la quinasa de unión SAPK/JNK1.3
Fue descrito por vez primera en 1970,4 y recientes estudios indican variantes alternativas del gen. Entre estas el exón novel en el intrón 8 nombrado exón 8B (8B FGD1) y el alelo 7B (7B FGD 1), que presenta un exón en el séptimo intrón.5,6 El 8B FGD 1 es expresado fuertemente en el cerebro, testes, médula espinal, tráquea y estómago, y débilmente en el timo y linfocitos.7 Sin embargo, la expresión del 7B FDG 1 es más débil y restringida a los testes y a las glándulas salivares.7 La inserción de cada exón novel conduce a la producción de una prematura terminación del codón, por lo que la proteína generada solo tiene dominios ricos en prolina y un incompleto dominio DH, que potencialmente compite con el gen salvaje FDG 1.7
El Síndrome de Aarskog se caracteriza por una fascie distintiva que se acompaña de baja talla y criptorquidia. El hallazgo de hipoplasia renal, anomalía poco frecuente, en dos de los pacientes diagnosticados de este síndrome, motivó que se profundizara en el estudio de su fenotipo, comparándolo con lo reportado por otros autores, ya podría tratarse de un nuevo genotipo, pendiente de corroborar mediante estudio molecular.
Se reportan los casos de 7 pacientes diagnosticados de síndrome de Aarskog en el servicio de Genética del Hospital Pediátrico Docente «William Soler». Se recogen los hallazgos fenotípicos referidos en sus historias clínicas y se comparan con los reportados en la literatura internacional.
PRESENTACIÓN DE LOS CASOS
Caso 1
Paciente varón de 4 años de edad, nacido a término, de parto eutócico, con un peso de 3030 g y talla de 50 cm. Entre los antecedentes patológicos familiares destacan la abuela materna con camptodactilia bilateral, la madre con pico de viuda e hipertelorismo y un tío abuelo con pico de viuda, referido por la madre del niño. Al momento de examen físico, a los 2 y medio años de edad, se encuentra talla de 71 cm (T/E entre 10mo y 25 percentilos).
El dismorfismo facial (figura 1) se recoge en la tabla 1. El desarrollo psicomotor fue normal, excepto por la demora en caminar hasta los 15 meses y el retraso en la erupción dentaria, con brote de un primer incisivo a los 11 meses. El ecocardiograma fue normal. Se detecta por ultrasonido (US) hipoplasia renal derecha. Presenta antecedentes de herniorrafia izquierda y orquidopexia bilateral. En la cistografía miccional se detectó reflujo vesico-ureteral de grado IV y en la gammagrafía, disminución del tamaño del riñón derecho. El test psicométrico mostró un coeficiente de inteligencia (CI) de 96.
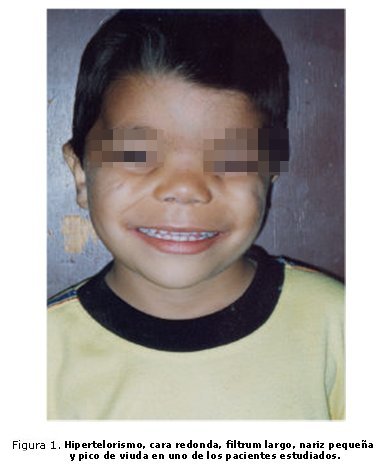
Tabla 1. Anomalías faciales
| Hallazgo | Pacientes | |||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | % | |
| Pico de viuda | + | + | + | + | + | + | + | 100,0 |
| Nariz corta | + | - | + | + | + | + | + | 85,7 |
| Puente nasal ancho | - | + | + | + | + | + | + | 85,7 |
| Filtro largo y ancho | + | + | + | - | + | + | + | 85,7 |
| Hipertelorismo | + | + | + | - | + | - | + | 71,4 |
| Frente amplia | - | + | + | + | + | + | - | 71,4 |
| Narinas antevertidas | + | + | + | - | - | + | + | 71,4 |
| Cara redonda | - | + | + | - | - | + | + | 57,1 |
| D. antimongoloide | - | + | + | - | + | - | - | 42,8 |
| Epicanto | + | + | - | - | - | - | + | 42,8 |
| Retraso en dentición | + | - | n.e | - | n.e | + | - | 40,0 |
| Cejas gruesas | - | + | + | - | - | - | - | 28,5 |
| Ptosis palpebral | - | + | - | - | + | - | - | 28,5 |
| Exotropia | + | - | - | - | - | - | - | 14,2 |
| Surco bajo labio inferior | - | - | - | - | + | - | - | 14,2 |
n.e: no explorado; +: anomalía presente; : anomalía ausente
Caso 2
Escolar del sexo masculino, de 4 años, nacido a término producto de parto eutócico, con peso de 2380 g y talla de 47 cm. El desarrollo psicomotor fue normal hasta el momento del examen. Los principales rasgos dismórficos se recogen en la tabla 1. En el examen radiográfico se halló un defecto costal y la escápula derecha elevada. El US mostró ectasia renal bilateral (riñón derecho 27 mm, izquierdo 15 mm). En el Ecocardiograma se observó válvula de Eustaquio redundante y un aneurisma de la fosa oval interatrial, sin defecto septal.
Caso 3
Madre del paciente anterior, de 37 años, que presenta al examen físico baja talla (143 cm), (T/E por debajo del 3er percentilo). Entre los rasgos fenotípicos se encontró pico de viuda, desviación antimongoloide de las hendiduras palpebrales, hipertelorismo ocular, puente nasal ancho, filtrum largo, cuello corto, así como miembros superiores e inferiores relativamente cortos y braquidactilia bilateral, más evidente en el 5to dedo de ambas manos.
Caso 4
Escolar de cinco años de edad, del sexo masculino, nacido a término de parto eutócico. Antecedentes maternos de ingestión de carbamazepina y fenobarbital para el tratamiento de la epilepsia durante el embarazo. Presentó hipoxia ligera al nacimiento. A los 2 meses fue operado de hernia inguinal. Sufrió de hepatitis, que ha recidivado en cuatro ocasiones. El cuadro dismórfico se refleja en la tabla 1. Impresiona pequeño (99 cm) para su edad (T/E entre 3er y 10mo percentilos).
Caso 5
Paciente de 67 años, abuelo materno de caso anterior. Las anomalías encontradas se recogen en la tabla 1. Los genitales no pudieron ser explorados.
Caso 6
Paciente de 2 años de edad, del sexo masculino, con antecedentes de parto por cesárea debido a posición transversa con peso de 3200 g y talla de 50 cm al nacimiento. Al momento del examen físico se encontró talla de 85 cm (entre 25 y 50 percentilos). Los rasgos dismórficos y malformaciones hallados se recogen en la tabla 1. Presentó retraso en la dentición y en el ecocardiograma se encontró una comunicación inter-atrial por fosa oval pequeña (2-3 mm), así como persistencia del conducto arterioso (PCA) en vías de cierre. El estudio renal por ultrasonido y el electroencefalograma practicado, fueron normales.
Caso 7
Paciente varón de 4 meses, con antecedentes prenatales de polihidramnios, macrosomía fetal y parto por cesárea debido a sufrimiento fetal agudo. El parto fue a término con peso 4050 g y talla adecuada. El dismorfismo facial se recoge en la tabla 1.
DISCUSIÓN
El rasgo dismórfico (tabla 1) más frecuente en los pacientes estudiados fue el pico de viuda (100 %). Las anomalías de los diferentes segmentos corporales (tabla 2) mas comunes fueron las extremidades cortas y la braquidactilia (figura 2 ), que también estuvieron presentes en todos los pacientes. El primero de los hallazgos dismórficos se observó también en el caso 2, correspondiente a una madre heterocigótica. Las extremidades cortas, de tipo rizomélico y la braquidactilia, predominaron sobre la baja talla (tabla 2). Mientras que algunos de los pacientes no están en los percentiles considerados como baja talla, otros han ido creciendo paralelamente a los percentiles inferiores, lo que se corresponde con lo reportado por otros autores.4,8
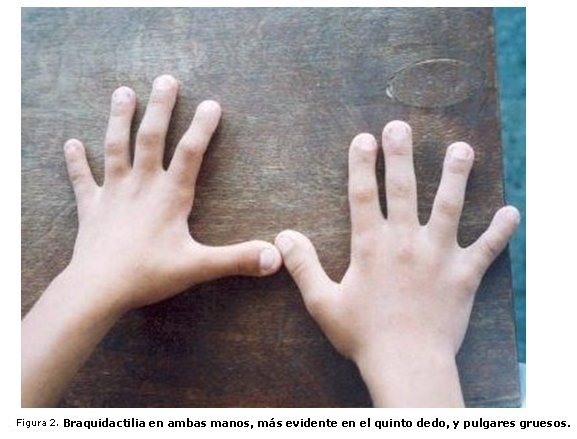
Tabla 2. Anomalías de talla y sistema osteomioarticular
| Hallazgos | Pacientes | % | ||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | ||
| Extremidades cortas | + | + | + | + | + | + | + | 100,0 |
| Braquidactilia | + | + | + | + | + | + | + | 100,0 |
| Cuello corto | - | + | + | + | + | - | + | 71,4 |
| Surco simiano | + | + | - | + | + | + | - | 71,4 |
| Clinodactilia 5to dedo | - | + | - | + | + | + | - | 57,1 |
| Baja talla | - | + | + | + | + | - | - | 57,1 |
| Pie varo | + | - | + | - | - | - | + | 42,8 |
| Cubito valgo | - | + | - | - | + | - | - | 28,5 |
| Pulgares anchos | + | - | - | - | + | - | - | 28,5 |
| Escoliosis | - | + | - | - | - | - | - | 14,2 |
| Asimetría de tórax | - | + | - | + | - | - | - | 14,2 |
| Pectus excavatum | - | + | - | - | - | - | - | 14,2 |
+: anomalía presente; : anomalía ausente
Otras rasgos faciales como la nariz corta, el puente nasal ancho, el filtrum largo, la frente amplia, las narinas antevertidas y el hipertelorismo (figura 2), resultaron más frecuentes que la disminución de las mensuraciones de determinados segmentos corporales, tales como la longitud del cuello y la criptorquidia (tabla 3), ya que, además de caracterizar al síndrome, son muy evidentes y llaman la atención del examinador. Las alteraciones de determinadas mensuraciones tales como la distancia interpupilar, la longitud del cuello, extremidades y las manos dependen de mediciones laboriosas y poco exactas, que muchas veces se estiman de modo subjetivo, lo que podría explicar la baja frecuencia encontrada, a pesar del rol que parece jugar la proteína FGD 1 en la morfogénesis y el crecimiento en general.3
Tabla 3. Anomalías genitourinarias
| Hallazgos | Pacientes | % | ||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | ||
| Criptorquidia | + | + | ---- | - | n.e | + | - | 60,0 |
| Hernia inguinal/abdominal | + | - | - | + | - | + | - | 42,8 |
| Escroto en chal | - | + | ---- | + | n.e | - | - | 40,0 |
| Escroto bífido | + | - | ---- | - | n.e | - | - | 20,0 |
| Riñones hipoplásicos | + | - | - | - | - | - | - | 14,2 |
| Reflujo vesico-ureteral | + | - | - | - | - | - | - | 14,2 |
| Pielectasia renal | + | + | - | - | - | - | - | 8,5 |
+: anomalía presente; anomalía ausente; n.e: no explorado; ——: no procede
Algunas de estos rasgos tales como la cara redonda, la micrognatia y el epicanto, sufren cambios con la edad, como se puede apreciar en los casos 3 y 5, que no presentan epicanto y en el paciente 5, en el que la cara es más bien alargada.9
Como se mencionó con anterioridad, otros hallazgos que definen el síndrome, entre estos, la criptorquidia, no fueron constantes en los pacientes estudiados. Este hecho se explicaría mediante los recientes avances en el estudio molecular, que indican claramente, que la variabilidad fenotípica esta determinada por la heterogeneidad alélica del síndrome. Pudiera especularse, que aquellas variantes con alteraciones genitales podrían tener el alelo 7B cuya competencia con el alelo salvaje produciría alteraciones de preferencia en testes y menos marcadas a otros niveles somáticos. Esta heterogeneidad pudiera también explicar, la presencia de hipoplasia renal en uno de los pacientes y las alteraciones pielo-caliciales y el reflujo en otros dos.
Existen mutaciones sin sentido encontrados en pacientes con retraso mental (RM) ligado al X no sindrómico, lo que sugiere la participación del producto del gen FDG1 en la génesis de algunos tipos de RM, o al menos, algunos de los dominios de esta proteína, son importantes en la cascada de eventos de la embriogénesis para el desarrollo normal del cerebro.3,10,11 No obstante, en los casos
estudiados no se detecto RM. Las anomalías cardiovasculares no fueron frecuentes en los pacientes estudiados al compararlas con otros hallazgos, pero si lo fueron con respecto a lo reportado hasta el momento.12
REFERENCIAS BIBLIOGRÁFICAS
2. A mutation in the pleckstrin homology (PH) domain of the FGD1 gene in an Italian family with faciogenital dysplasia (Aarskog-Scott syndrome). [Citado en 2005]. Disponible en: http://www.ncbi.nhl.nih.gov/entrez/query.fcgi
3. The Caenorhabditis elegans homolog of FGD1, the human Cdc42 GEF gene responsible for faciogenital dysplasia, is critical for excretory cell morphogenesis. Hum Mol Genet. [Citado en 2001]. Disponible en: http://www.ncbi.nhl.nih.gov/entrez/query.fcgi
4. Aarskog D. A familial syndrome of short stature associated with facial dysplasia and genital anomalies. J. Pediatr 1970; (77):856.
5. Yi Chuan Xue Bao. Analysis, identification and correction of some errors of model reference seqments appeared in NCBI Human Gene Database by in silico cloning and experimental verification of novel human genes. [Citado en 2005]. Disponible en: http://www.ncbi.nhl.nih.gov/entrez/query.fcgi
6. Skeletal-specific expression of FDG 1 during bone formation and skeletal defects in faciogenital dysplasia (FGDY; Aarskog syndrome). [Citado en 2000]. Disponible en: http://www.ncbi.nhl.nih.gov/entrez/query.fcgi
7. Yanagi K, Kaname T, Chinen Y, Naritomi K. Novel alternative splicing of human faciogenital dysplasia 1 gene. [Citado en 2005]. Disponible en: http://www.ncbi.nhl.nih.gov/entrez/query.fcgi
8. Oberiter V, Lovrencic MK, Schmutzer L, Kraus O. The Aarskog syndrome. Acta Paediatr Scand 1980; 69 (4):567-70.
9. Fryns JP. Aarskog syndrome. The Changing phenotype with age. Am J Med Genet 1992; (43):420.
10. Orrico A., Galli L, Buoni S, Hayek G, Luchetti A, Lorenzini S, et al. Attention-deficit/hyperactivity disorder (ADHD) and variable clinical expression of Aarskog-Scott syndrome due to a novel FGD1 gene mutation (R408Q). Am J Med Genet 2005; (35):99-102.
11. Lebel. Non-syndromic X-linked mental retardation associated with a missense mutation (P312L) in the FGD1 gene. Clin Genet 2002; (61):139-145.
12. Fernandez I, Tsukahara M, Mito H, YoshiiH, Uchida M, Matsuo K, et al. Congenital heart defects in Aarskog syndrome. Am. J. Med. Genet 1994; 50: 318-322.
Recibido: 23 de abril de 2008.
Aprobado: 16 de julio de 2008.
Iván Hernández García. San Francisco y Perla, Altahabana. La Habana, Cuba.
Correo electrónico:debora.garcia@infomed.sld.cu

