










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr v.81 n.2 Ciudad de la Habana abr.-jun. 2009
Aspectos clínicos y electroencefalográficos de la epilepsia focal en el niño
Clinical and electroencephalographic features of focal epilepsy present in children
Albia Pozo Alonso,I Desiderio Pozo Lauzán,II Maritza Oliva Pérez III
I Especialista de II Grado en Pediatría y Neurología. Profesora Auxiliar. Investigadora Auxiliar. Hospital Pediátrico Docente «William Soler». La Habana, Cuba.
II Especialista de II Grado en Neurología y Pediatría. Doctor en Ciencias Médicas. Profesor Titular y Consultante. Hospital Pediátrico Docente «William Soler». La Habana, Cuba.
III Especialista de II Grado en Bioestadística. Profesora Auxiliar. Policlínico Docente «Salvador Allende». La Habana, Cuba.
RESUMEN
INTRODUCCIÓN. El objetivo del presente trabajo fue caracterizar un grupo de pacientes con epilepsias focales, según aspectos clínicos y electroencefalográficos.
MÉTODOS. Se realizó un estudio descriptivo y prospectivo de 185 niños con diagnóstico de epilepsia focal (2 o más crisis epilépticas no provocadas), con edades entre un mes y 14 años, que fueron hospitalizados en el Departamento de Neuropediatría del Hospital «William Soler» entre diciembre de 2001 y diciembre de 2003.
RESULTADOS. La edad media de inicio de la primera crisis epiléptica fue de 5 años. El tipo de crisis epiléptica focal más frecuente fue la simple (49,2 %). El 48,6 % de los niños presentó etiología idiopática y el 33,0 % sintomática. Los factores de la etiología sintomática más frecuentes fueron los prenatales (56,2 %). El 91,4 % de los pacientes presentó electroencefalogramas iniciales interictales anormales. El electroencefalograma focal se observó en el 37,3 % de los niños y el multifocal en el 24,9 %. El síndrome epiléptico más frecuente fue la epilepsia benigna con puntas centrotemporales (5,9 %).
CONCLUSIONES. Los niños con epilepsia focal tienen variadas manifestaciones clínicas y electroencefalográficas, y en la mayoría de los pacientes no es posible identificar un síndrome epiléptico.
Palabras clave: Epilepsias focales, síndromes epilépticos focales, crisis epilépticas focales.
ABSTRACT
INTRODUCTION: The aim of present paper was to characterize a group of patients presenting with focal epilepsies by clinical and electroencephalographic features.
METHODS: Authors made a descriptive and cross-sectional study in 185 children diagnosed with focal epilepsy (two or more non-provoked epilepsy crises), aged from one month to 14, admitted in Neurology Department of "William Soler" Children Hospital between December 2001 to December 2003.
RESULTS: Mean age of the first epilepsy crisis was at 5 years. The more frequent type of focal epilepsy crisis was the simple one (49, 2%). The 48, 6% of children presented with a idiopathic origin, and the 33, 0% if symptomatic one. The more frequent factors of symptomatic origin were the prenatal ones (56, 2%). The 91, 4% of patients had abnormal interictal initial electroencephalograms. The focal electroencephalogram was observed in the 37, 3% of children, and the multifocal one in the 24, 9%. The more frequent epileptic syndrome was the benign epilepsy with central-temporal waves (5, 9%).
CONCLUSIONS: Children presenting with epilepsy have many clinical and electroencephalographic manifestations and in most of patients it is not possible to identify an epilepsy syndrome.
Key words: Focal epilepsies, focal epilepsy syndromes, focal epilepsy crisis.
INTRODUCCIÓN
En las crisis epilépticas focales la semiología inicial indica la activación inicial de solamente parte de un hemisferio cerebral.1-4
Las crisis focales llegan a constituir hasta el 49,3 % de las epilepsias en la edad escolar y la adolescencia; son el tipo de mayor prevalencia entre las epilepsias.5 Sin embargo, algunos autores6,7 consideran que las epilepsias focales son poco frecuentes durante los primeros 3 años de la vida, sobre todo debido a que resulta difícil su identificación en el lactante.
Estudios de monitorización de video-electroencefalograma realizados en lactantes con crisis epilépticas han mostrado que con frecuencia es difícil distinguir las crisis focales de las generalizadas, sobre la base de la observación clínica y de los electroencefalogramas (EEG) interictales.8
Las epilepsias focales consisten en la repetición de crisis epilépticas que implican una localización cortical focal. No todas las epilepsias focales pueden incluirse en un síndrome epiléptico.2,9
Para el diagnóstico de un síndrome epiléptico se deben considerar varios aspectos como la edad de comienzo de las crisis epilépticas, tipo de crisis epiléptica, historia de salud del paciente, causa, factores precipitantes, hallazgos al examen neuropsicológico, hallazgos electroencefalográficos y estudios de neuroimagen.4,9
A través de los años, la epilepsia ha sido el trastorno más frecuente observado en el Departamento de Neuropediatría del Hospital Pediátrico Docente «William Soler». Entre las epilepsias, las focales son la causa más frecuente de consulta y hospitalización en dicho departamento. El objetivo de la presente investigación fue caracterizar un grupo de pacientes con epilepsias focales según aspectos clínicos y electroencefalográficos.
MÉTODOS
Se realizó un estudio descriptivo y prospectivo en el Departamento de Neuropediatría del Hospital «William Soler». El universo de estudio estuvo constituido por todos los niños entre un mes y 14 años de edad que presentaron dos o más crisis epilépticas focales no provocadas por una causa de identificación inmediata (epilepsia focal), hospitalizados en el mencionado departamento entre diciembre de 2001 y diciembre de 2003. Fueron estudiados un total de 185 niños. Se excluyeron del estudio los pacientes que presentaron crisis epilépticas agudas de causa extracraneal o intracraneal, crisis epilépticas generalizadas, crisis epilépticas febriles, crisis cerebrales anóxicas reflejas (vagales) y crisis cerebrales psíquicas.
Se efectuaron evaluaciones del neurodesarrollo y del coeficiente de inteligencia y se aplicó, de acuerdo a la edad del paciente en el momento del diagnóstico, el Manual para las Escalas de Desarrollo Infantil de Nancy Bayley,10 en los menores de 3 años, y el Test de Terman-Merrill,11 en los de 3 a 14 años. Se aplicó la clasificación de retraso mental de acuerdo con la Asociación Americana de Psiquiatría, del año 2000.12
A todos los pacientes se les realizó un registro electroencefalográfico (EEG) al inicio del diagnóstico. En el 56 % de los casos se realizó de vigilia; en el 30,2 %, de sueño y en el 13,8 % de vigilia y sueño. En el 28,8 % de los pacientes en los que se obtuvieron trazados electroencefalográficos de sueño, el estudio se realizó con privación de sueño de 24 h en los mayores de 5 años. En el 26,2 % se realizó con privación parcial de 5 h; en el 30 %, de sueño espontáneo y en el 15 % se obtuvo el sueño artificial o inducido con la administración de hidrato de cloral en dosis de 50 mg/kg, como dosis máxima, administrada por vía oral.
En los registros de vigilia se efectuaron las maniobras siguientes: apertura y cierre de los ojos, hiperventilación por vía bucal durante 3 min y fotoestimulación.
Se utilizó un electroencefalógrafo de 14 canales modelo 7414K y otro de 8 canales modelo 7501. Se utilizaron diferentes montajes con el objetivo de obtener la actividad eléctrica de todas las regiones cerebrales. La colocación de los electrodos se realizó de acuerdo con el Sistema Internacional 10-20.13
En correspondencia con la valoración clínica se realizó tomografía axial computarizada de cráneo simple o contrastada (TAC) en el 70,2 % de los pacientes, imagen de resonancia magnética de cráneo (IRM) en el 9,2 % de los niños, tomografía por emisión de positrón simple (SPECT), cariotipo, hemograma, gasometría, ácido láctico, amonio, pruebas metabólicas en orina, cromatografía de aminoácidos en sangre y orina, estudio enzimático lisosomal en leucocitos y otras determinaciones.
Las variables utilizadas fueron las siguientes:
- Edad: Se consideró la edad de comienzo de la primera crisis epiléptica no provocada. Se situó entre 1 mes y 14 años y se distribuyó de la forma siguiente: 1 mes hasta 11 meses, de 1 año hasta 4 años, de 5 a 9 años y de 10 a 14 años.
- Sexo: Masculino y femenino.
- Tipo de crisis epiléptica: El tipo de crisis epiléptica focal se consideró de acuerdo con la clasificación internacional de las crisis epilépticas, de 1981.3
- Etiología de la epilepsia: Se consideraron las etiologías siguientes:14
- Idiopática: Pacientes con ausencia de lesión cerebral subyacente, examen neurológico, intelecto normal y predisposición genética.
- Sintomática: Pacientes en los que se pudo precisar la causa de la epilepsia.
- Probablemente sintomática (criptogénica): Pacientes con anomalías en el examen neurológico y discapacidades neurológicas asociadas en los que no se pudo precisar la causa de la epilepsia.
- Factores de la etiología sintomática: Dentro de la etiología sintomática se tuvieron en cuenta los factores prenatales, perinatales y posnatales. Se consideraron crisis neonatales sintomáticas las que ocurrieron antes de los 28 días de vida, provocadas por diferentes causas. Se consideraron antecedentes personales de crisis febriles los que ocurrieron en niños con edades entre 3 meses y 5 años, asociadas con fiebre de 38,5 ºC o más, en ausencia de infección del sistema nervioso central.15 Los antecedentes familiares de epilepsia se consideraron teniendo en cuenta a los familiares de primer grado (padre, madre o hermanos) y a los familiares de segundo grado (tíos, primos y abuelos). Como discapacidades neurológicas asociadas se tomaron en cuenta las siguientes: parálisis cerebral, retraso del neurodesarrollo global y retraso mental.
- Estado epiléptico: Se consideró a las crisis epilépticas con una duración mayor de 30 min y a las crisis epilépticas consecutivas entre las que no hay recuperación de la conciencia.
- Parálisis de Todd: Se tuvo en cuenta a los pacientes que presentaron disfunción postictal unilateral relacionada con funciones motoras y somatosensitivas.
- EEG inicial interictal: Se clasificó como normal o anormal y en este último caso se describieron los hallazgos electroencefalográficos. Los electroencefalogramas se consideraron normales de acuerdo con diferentes parámetros de los trazados de vigilia y de sueño.13 Los trazados electroencefalográficos anormales que se consideraron fueron:
- focal, cuando las alteraciones estaban localizadas en una o dos áreas diferentes, en un hemisferio o en ambos;
- multifocal, si las alteraciones del trazado estaban localizadas en más de 2 áreas corticales, limitadas a uno o ambos hemisferios;
- en un hemisferio, cuando las descargas se localizaron en un hemisferio;
- generalizado, si las alteraciones electroencefalográficas se observaron en todas las áreas corticales en forma sincrónica;
- focal que se generaliza, cuando las alteraciones focales se propagaron a todas las áreas corticales;
- multifocal que se generaliza, si las alteraciones multifocales se propagaron a todas las áreas corticales;
- en un hemisferio que se generaliza: las alteraciones localizadas en un hemisferio cerebral se propagaron al hemisferio contralateral.
- Síndrome epiléptico: Se consideraron de acuerdo con el esquema diagnóstico propuesto por la Liga Internacional contra la Epilepsia, en el 2001.14
Se construyeron distribuciones de frecuencias absolutas y relativas. Como medidas de resumen se emplearon el porcentaje y la media con la desviación estándar según el tipo de variables cualitativa o cuantitativa, respectivamente.
RESULTADOS
Al sexo masculino correspondió el 56,8 % de los pacientes. El grupo de edad más frecuente fue el de 5 a 9 años (37,3 %), seguido por el de 1 a 4 años (35,7 %). El grupo de edad menos frecuente fue el de 10 a 14 años (12,4 %). El grupo de menores de un año estuvo constituido por el 14,6 % de los casos. La edad media de la primera crisis focal fue de 5,0 años con una desviación estándar de 3,84 años.
En la tabla 1 se aprecia que el tipo de crisis focal más frecuente fue la simple, la cual se observó en prácticamente la mitad de los pacientes. El tipo de crisis secundariamente generalizada se constató en las dos quintas partes de los niños. Las crisis focales complejas se evidenciaron en el 8,6 % de los pacientes y la simple-compleja, en el 1,6 %.
Tabla 1. Tipo de crisis epilépticas focales
| Crisis epilépticas focales | n | % |
| Simple | 91 | 49,2 |
| Secundariamente generalizada | 75 | 40,5 |
| Compleja | 16 | 8,7 |
| Simple-compleja | 3 | 1,6 |
| Total | 185 | 100 |
Las crisis epilépticas iniciales se presentaron en vigilia en el 69,2 % de los pacientes. El estado epiléptico se registró en el 5,9 %. El 72,7 % presentó una etiología sintomática; el 18,2 %, idiopática y el 9,1 %, criptogénica. El 72,7 % de los pacientes inició el estado de mal epiléptico a la edad de 3 años o menos. Las crisis epilépticas más frecuentes en los pacientes que presentaron estado epiléptico fueron las focales secundariamente generalizadas (72,7 %) y las focales simples motoras (18,2 %). La parálisis de Todd se observó en 13 niños (7,0 %).
El antecedente personal más frecuente en el total de pacientes fueron las discapacidades neurológicas que se evidenciaron en el 30,3 %. El 14,6 % del total de pacientes presentó parálisis cerebral; el 13,5 %, retraso mental y el 11,9 %, retraso del neurodesarrollo global.
Los antecedentes personales de crisis neonatales sintomáticas estuvieron presentes en el 10,8 % y las crisis febriles previas en el 9,7 %.
El antecedente patológico familiar de epilepsia se observó en el 33,5 % de los pacientes incluidos en el estudio.
La etiología más frecuente fue la idiopática, la cual se observó en 90 pacientes (48,6 %). La etiología sintomática se observó en 61 niños (33,0 %) y la criptogénica se evidenció en 34 (18,4 %). Dentro de la etiología sintomática los factores prenatales fueron los más frecuentes y se presentaron en el 60,7 % de los pacientes de forma individual, y combinados con otros factores (perinatales y posnatales) en el 8,2 %, y presentes en total en el 68,9 % de los pacientes con esta etiología. Los factores perinatales se evidenciaron en el 18 % y los posnatales en el 13,1 %.
El factor prenatal más frecuente fue la atrofia cerebral de cualquier localización, la cual afectó a más de la mitad de estos pacientes. La prematuridad sola o asociada a otra patología se observó en el 13,5 % de los niños y la esclerosis del hipocampo, la poroencefalia y el síndrome de Sturge-Weber se evidenciaron en el 5,4 % de los casos. El resto de las causas prenatales se presentaron en un caso cada una (tabla 2).
Tabla 2. Factores etiológicos prenatales
| Factores prenatales | n | % |
| Atrofia cortical o de hipocampo * | 20 | 54,1 |
| Prematuridad sola o asociada a otra patología | 5 | 13,5 |
| Esclerosis del hipocampo | 2 | 5,4 |
| Poroencefalia | 2 | 5,4 |
| Síndrome de Sturge-Weber | 2 | 5,4 |
| Esclerosis tuberosa | 1 | 2,7 |
| Atrofia del hipocampo-heterotopia focal | 1 | 2,7 |
| Agenesia del cuerpo calloso | 1 | 2,7 |
| Eclampsia-infarto cerebral | 1 | 2,7 |
| Leucoencefalodisplasia-eclampsia | 1 | 2,7 |
| Placenta previa-infarto cerebral | 1 | 2,7 |
| Total | 37 | 100 |
* Cinco pacientes presentaron atrofia del hipocampo.
El factor perinatal más frecuente fue la asfixia, la cual se observó en el 81,8 % de los pacientes con este factor. En el 9,1 % de los niños se evidenció asfixia asociada con hipoglucemia y broncoaspiración, respectivamente.
Los factores posnatales más frecuentes fueron la sepsis neonatal y la meningoencefalitis bacteriana que se observaron en el 37,5 % de los pacientes respectivamente. La encefalitis por herpes virus y el paro cardiorrespiratorio estuvieron presentes en un paciente cada uno.
El EEG se realizó en todos los pacientes estudiados. La mayoría de los pacientes 169 (91,4 %) presentó electroencefalogramas iniciales interictales anormales. De ellos, el 37,3 % tuvo EEG focales, el 24,9 % EEG multifocales y el 15,3 % mostró EEG multifocales que se generalizaron (tabla 3) (figura).
Tabla 3. Electroencefalogramas iniciales interictales anormales
| Electroencefalogramas iniciales | n | % |
| Focal | 63 | 37,3 |
| Multifocal | 42 | 24,9 |
| Multifocal-generalizado | 26 | 15,3 |
| Focal-generalizado | 20 | 11,8 |
| Generalizado | 8 | 4,7 |
| Un hemisferio-generalizado | 6 | 3,6 |
| Un hemisferio | 4 | 2,4 |
| Total | 169 | 100 |
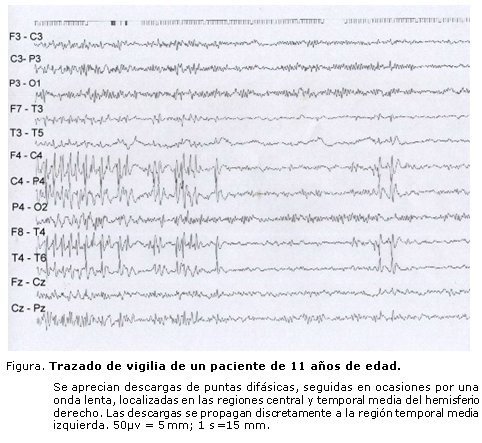
La tabla 4 muestra que sólo en 31 pacientes (16,8%) se identificaron síndromes epilépticos focales. El síndrome epiléptico más frecuente fue la epilepsia benigna con puntas centrotemporales (6,0%), las crisis infantiles benignas no familiares se observaron en el 2,7% y la epilepsia occipital benigna de comienzo precoz en el 2,2%. Los síndromes menos frecuentes observados fueron la epilepsia benigna con sintomatología afectiva, el síndrome hemiconvulsión-hemiplejía y las crisis parciales migratorias de la lactancia que se evidenciaron en el 0,5% de los niños.
Tabla 4. Síndromes epilépticos focales
| Síndromes epilépticos | n | % | |
| Idiopáticos | Crisis infantiles benignas no familiares | 5 | 2,7 |
| Epilepsia benigna con puntas centrotemporales | 11 | 6,0 | |
| Epilepsia occipital benigna de comienzo precoz | 4 | 2,2 | |
| Epilepsia occipital de comienzo tardío (tipo Gastaut) | 2 | 1,1 | |
| Crisis parciales benignas de la adolescencia * | 2 | 1,1 | |
| Epilepsia benigna con sintomatología afectiva * | 1 | 0,5 | |
| Sintomáticos | Epilepsia del lóbulo temporal mesial con esclerosis del hipocampo | 2 | 1,1 |
| Síndrome de Sturge-Weber | 2 | 1,1 | |
| Síndrome hemiconvulsión-hemiplejía | 1 | 0,5 | |
| Crisis parciales migratorias de la lactancia | 1 | 0,5 | |
| Subtotal | 31 | 16,8 | |
| No síndromes | 154 | 83,2 | |
| Total | 185 | 100,0 | |
* Síndromes no incluidos en el esquema diagnóstico del año 2001.
DISCUSIÓN
En el estudio de Jiménez-Parrilla y otros16 la mayoría de los niños tuvieron el inicio de las crisis focales a la edad de 5,7 años. En nuestro trabajo la edad de inicio de las crisis epilépticas fue más frecuente en el grupo de 5 a 9 años (37,3 %). La edad media fue de 5,0 años, lo que indica un inicio temprano de las crisis epilépticas focales.
Jiménez-Parrilla y cols.16 observaron en su serie que la mayoría de las crisis fueron secundariamente generalizadas (47,8 %), seguidas por las crisis focales simples en el 36 % de los casos y en el 16,2 % se trataba de crisis focales complejas. A diferencia de estos autores, en nuestro estudio predominaron las crisis focales simples.
Consideramos que el tipo de crisis epiléptica que presentan los pacientes es un factor controvertido. Se ha comprobado17 que existe una gran variabilidad entre diferentes observadores al aplicar la Clasificación Internacional de las crisis epilépticas,3 lo que dificulta la comparación de unos estudios con otros.
A diferencia de nuestro estudio, Shinnar y otros18 encontraron en su serie de niños con epilepsias focales que la etiología más frecuente fue la sintomática (47,4 %). La etiología criptogénica se observó en el 29,8 % y la idiopática en el 22,8 %.
La atrofia cerebral es una lesión del sistema nervioso central (SNC) causada por una pérdida irreversible de la sustancia cerebral, y puede originarse por la ocurrencia de diferentes procesos a ese nivel: infartos, inflamación, desmielinización, hipoxia, o puede ser la consecuencia en el SNC de traumas, radioterapia, quimioterapia.19 También se ha planteado que se debe a disgenesias cerebrales prenatales.20
En nuestro trabajo se consideró que la atrofia cerebral fue de causa prenatal en los pacientes en los que no encontraron otras causas que explicaran la presencia de atrofia cerebral como las ya mencionadas. Se debe enfatizar en que la atrofia y la esclerosis del hipocampo evidenciadas en nuestros pacientes se consideraron también de origen prenatal, debido a microdisgenesias hipocampales porque no se encontraron otros factores causales como antecedentes personales de crisis febriles,21-23 traumatismos craneoencefálicos,21 infecciones del SNC,21,24 crisis epilépticas frecuentes,24 y prolongadas.25 En realidad, el poder precisar si la atrofia del hipocampo y la esclerosis del hipocampo son causa o consecuencia de la epilepsia es un aspecto muy debatido en la literatura médica en la actualidad.22,26
En nuestro estudio, predominó entre las discapacidades neurológicas la parálisis cerebral, seguida por el retraso mental y el retraso global del neurodesarrollo. El 26,7 % de los pacientes de Jiménez-Parrilla y cols.16 diagnosticados de epilepsias focales presentaron retraso global del neurodesarrollo.
En el estudio de Sillanpää y otros,27 el retraso mental se observó en el 33 % de los casos y la parálisis cerebral ocurrió en el 17 %. Este autor tomó en consideración a los pacientes con epilepsias focales y generalizadas.
Jiménez-Parrilla y otros16 observaron que el 20 % de sus pacientes presentó EEG normales iniciales y encontraron anomalías paroxísticas focales en el 41,8 %, multifocales en el 16,2 % y alteraciones focales que se generalizaron con posterioridad en el 5,8 %. Nuestros hallazgos coincidieron con estos autores porque la mayoría de nuestros niños presentó EEG interictales iniciales anormales y predominaron los trazados focales y multifocales.
En la presente investigación los síndromes epilépticos focales se clasificaron de acuerdo con el esquema propuesto para personas con crisis epilépticas y epilepsia de la Liga Internacional contra la Epilepsia, en el año 2001,14 porque se incluyen más síndromes que en la clasificación vigente de las epilepsias y síndromes epilépticos del año 1989.9 Incluso, se incluyeron dos síndromes que no aparecen en el esquema propuesto del año 2001.14 A pesar de ello, no se logró la identificación de los síndromes epilépticos en la mayoría de los pacientes.
Varios autores plantean una alta proporción de niños epilépticos que no reúnen los criterios de síndromes epilépticos específicos.28,29
Se concluye que los niños con epilepsias focales tienen variadas manifestaciones clínicas y electroencefalográficas. La mayoría de las epilepsias focales no se incluyen en síndromes epilépticos.
REFERENCIAS BIBLIOGRÁFICAS
1. Blume WT, Luders H, Mizrahi E, Tassinari C, van Em de Boas W, Engel J, Jr. Glossary of descriptive terminology for ictal semiology: report of the ILAE. Task Force on Classification and Terminology. Epilepsia 2001;42:1212-18.
2. Pozo Alonso A, Pozo-Lauzán D, Pozo-Alonso D. Síndromes epilépticos parciales idiopáticos. Rev Neurol 2001;33:1064-70.
3. Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsia 1981; 22:489-501.
4. Pozo Lauzán D, Pozo A. Epilepsias y discapacidades neurológicas en el niño. La Habana: Editorial de Ciencias Médicas, 2007.
5. Rufo-Campos M. Crisis parciales en la infancia. Rev Neurol 2001;32:962-9.
6. Oller-Daurella L, Oller LF. Partial epilepsy with seizures appearing in the first three years of life. Epilepsia 1989;30:820-6.
7. Chakova L. Studies on the frecuency and clinical manifestations of epilepsy in infancy and early childhood. Folia Medica 1996;38:75-80.
8. Nordi DR Jr, Bazil CW, Scheuer ML, Pedley TA. Recognition and classification of seizures in infants. Epilepsia 1997;38:553-60.
9. Commission on classification and terminology of the international league against epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989;30:389-99.
10. Bayley N. Manual for the scales of infant development. New York: the Psychological Corporation, 1986.
11. Terman LM, Merrill MA. Measuring Intelligence. Boston: Houghton Mifflin, 1937.
12. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th ed. Washington, DC: American Psychiatric Association, 2000.
13. Crespel A, Gélisse P. Atlas d'électroencéphalographie. Tome I. Paris: John Libbey Eurotext, 2005.
14. Engel J, Jr. A proposed diagnostic scheme for people with epileptic seizures and with epilepsy. Report of the ILAE Task Force on Classification and Terminology. Epilepsia 2001;42:1-8.
15. Camfield P, Camfield C. Les crises fébriles. En: Roger J, Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P, Thomas P, Hirsch E, eds. Les syndromes épileptiques de l'enfant et de l'adolescent. 4ème edn. Montrouge: John Libbey Eurotext Ltd, 2005:159-70.
16. Jiménez-Parrilla F, López-Corona JM, Vázquez-Florido AM, Madruga-Garrido M, Ruiz del Portal L, Rufo-Campos M. Eficacia de los fármacos antiepilépticos en el tratamiento de las crisis parciales en la infancia. Análisis comparativo de 90 casos. Rev Neurol 2001;33:250-91.
17. Bodensteiner JB, Brownsworth RD, Knapik JR, Kanter MC, Cowan LD, Leviton A. Interobserver variability in the ILAE classification of seizures in childhood. Epilepsia. 1988;29:123-8.
18. Shinnar S, O'Dell C, Berg A. Distribution of epilepsy syndromes in a cohort of children prospectively monitored from the time of their first unprovoked seizures. Epilepsia 1999; 40: 1378-83.
19. Pérez E, González I, Noda M, González E, Cabal C, Losada J. Atrofia cortical en pacientes con anemia de células falciformes. Estudio por imágenes de resonancia magnética. Rev Neurol 2001;32:1192-93.
20. Ohtahara S, Ohtsuka Y, Yamatogi Y, Oka E, Inoue H. L'encéphalopathie épileptique infantile précoce avec "suppression-burst". En: Roger J, Bureau J, Dravet C, Dreifuss FE, Perret A, Wolf P, eds. Les syndromes épileptiques de l'enfant et de l'adolescent. 2ème edn. London: John Libbey & Company Ltd, 1992: 25-34.
21. Zamecnik J, krsek P, Marusic P, Druga R, Benes V, Tichy M, komarek V. Microscopic disorders of cortical development of the brain and its etiopathogenic importance for detection in patients with temporal epilepsy associated with hippocampal sclerosis. Cesk Patol 2003;39:178-84.
22. Janszky J, Woermann FG, Schulz R, Halász P, Ebner A. Right hippocampal sclerosis is more common than left after febrile seizures. Neurology 2003;60:1209.
23. Brandão EM, de Manreza ML. Mesial temporal sclerosis in children. Arq Neuropsiquiatr 2007;65:947-50.
24. Salmenperä T, Könönen M, Roberts N, Vanninen R, Pitkänen A, Kälviäinen R. Hippocampal damage in newly diagnosed focal epilepsy. A prospective MRI study. Neurology 2005;64:162-8.
25. Chevret L, Husson B, Nguefack S, Nehlig A, Bouilleret V. Prolonged refractory status epilepticus with early and persistent restricted hippocampal signal MRI abnormality. J Neurol 2008; 255:112-6.
26. Yu-tze NG, Mc Gregor AL, Wheless JW. Magnetic resonance imaging detection of mesial temporal sclerosis in children. Pediatric Neurology 2004;30:81-5.
27. Sillanpää M. Long-term prognosis in finish childhood-onset epilepsy. En: Jallon P, Berg A, Dulac O, Hauser A, eds. Prognosis of epilepsies. Paris: John Libbey Eurotext, 2003:127-34.
28. Berg AT. Identification of epilepsy syndromes at diagnosis and modification with time. En: Jallon P, Berg A, Dulac O, Hauser A, eds. Prognosis of epilepsies. Paris: John Libbey Eurotext. 2003;185-96.
29. Jallon P, Loiseau J. Newly diagnosed unprovoked epileptic seizures: presentation at diagnosis in CAROLE study. Epilepsia 2001;42:464-75.
Recibido: 29 de enero de 2009.
Aprobado: 16 de marzo de 2009.
Albia Pozo Alonso. Hospital Pediátrico Docente «William Soler». Departamento de Neuropediatría. Calle 100 y Perla. Altahabana. CP 10800. Boyeros. La Habana, Cuba.
Correo electrónico: albiap@infomed.sld.cu

