










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr v.81 n.3 Ciudad de la Habana jul.-sep. 2009
Tetralogía de Fallot y artrogriposis
Tetralogy of Fallot related to arthrogryposis
Jacqueline Barrial Moreno,I Norma Elena de León Ojeda,II Eugenio Selman-Housein Sosa,III Maria Teresa Consuegra Chuairey,IV Gilberto Bermudez Gutiérrez V
I Especialista de I Grado en Anestesiología y Reanimación. Máster en Urgencias Médicas. Cardiocentro Pediátrico «William Soler». Instructor del Departamento de Cirugía, Facultad de Ciencias Médicas «Enrique Cabrera». La Habana, Cuba.
II Especialista de II Grado en Genética Clínica. Máster en Bioética. Hospital Pediátrico «William Soler». Asistente del Departamento de Genética Médica del ICBP Victoria de Girón. La Habana, Cuba.
III Doctor en Ciencias Médicas. Especialista de II Grado en Cardiocirugía Pediátrica. Profesor Titular. Cardiocentro Pediátrico «William Soler». La Habana, Cuba.
IV Especialista de I Grado en Pediatría. Instructora. Cardiocentro Pediátrico «William Soler». La Habana, Cuba.
V Especialista de I Grado en Cirugía Cardiovascular Pediátrica. Cardiocentro Pediátrico «William Soler». La Habana, Cuba.
RESUMEN
La tetralogía de Fallot es una cardiopatía cianótica que representa el 5,4 % de todos los defectos cardíacos congénitos. El 12 % de estos pacientes pueden tener anomalías cromosómicas asociadas. Se presenta el caso de paciente, nacido de un parto gemelar, con edad de 10 meses y peso corporal de 8 kg, que nació con tetralogía de Fallot y artrogriposis distal. Se describe el manejo anestésico transoperatorio así como la evolución posoperatoria y las complicaciones que se presentaron.
Palabras clave: Tetralogía de Fallot, artrogriposis, evolución posoperatoria y complicaciones.
ABSTRACT
Tetralogy of Fallot is a cyanotic heart disease representing 5,4% of all the congenital cardiac defects. The 12% of these patients may to have associated chromosomal anomalies. Authors present the case of patient born of twins labor aged 10 months and a body weight of 8 k born with Tetralogy of Fallot and distal arthrogryposis. We describe transoperative anesthetetic management as well as postoperative course and complications present.
Key words: Tetralogy of Fallot, arthrogryposis, postoperative course, and complications.
INTRODUCCIÓN
Según la definición actual, el término artrogriposis se refiere a la existencia de más de una zona del cuerpo afectada por contracturas congénitas, lo cual se observa en 1/3000 nacidos vivos.1 Los defectos congénitos de este tipo son anomalías anatómicas o funcionales presentes al nacimiento o después de este, y reconocibles al examen clínico.1-3 La limitación del movimiento articular condiciona la aparición de la artrogriposis congénita (contracturas congénitas), que puede presentarse aislada o distal y múltiple. Las distales comprenden varios tipos entre las que se encuentra la artrogriposis distal de tipo II o displasia digitotalar, entidad autosómica dominante. La gemelaridad puede condicionar la discordancia entre gemelos y está reportada la artrogriposis múltiple o amioplasia congénita en uno de los gemelares.4,5
La tetralogía de Fallot es un defecto cardíaco congénito cianótico, relativamente frecuente y caracterizado por una comunicación interventricular acompañada de dextroposición de la aorta, obstrucción del tracto de salida del ventrículo derecho que presenta hipertrofia muscular.6 En este trabajo se presenta el caso de un paciente con artrogriposis distal y tetralogía de Fallot, que fue operado para corrección de su cardiopatía.
PRESENTACIÓN DEL CASO
Paciente varón, gemelo dicigótico, de 10 meses de edad y 8 kg de peso, hijo de padres no consanguíneos, sin defectos en extremidades, con hermano sin defectos congénitos, nacido por parto eutócico a término, y remitido de su provincia para tratamiento de una tetralogía de Fallot. Al examen físico presentaba contracturas congénitas en los miembros inferiores, con pie varoequino bilateral, con hoyuelos periarticulares, desviación cubital de los dedos de las manos con apariencia de camptodactilia pero reductibles al examen físico y hendidura en la porción media del mentón, que recordaba la barbilla en H (figura). Fue evaluado por el genetista clínico que planteó una artrogriposis distal de tipo II. No presentaba anomalías en la columna cervical que dificultaran su movilidad. El paciente tenía cianosis moderada, y se confirmó el diagnóstico de cardiopatía y sus características anatómicas por ecocardiografía. La radiografía de tórax y el electrocardiograma (ECG) resultaron característicos de la tetralogía de Fallot.
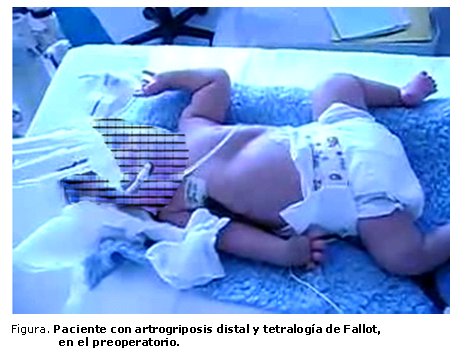
Una vez en el quirófano, se premedicó por vía intramuscular con ketamina (6 mg/kg) y atropina (0,02 mg/kg) para separarlo de sus familiares sin provocar situaciones de estrés (llanto, irritabilidad). Se crearon las condiciones dentro del quirófano para evitar hipotermia en un niño pequeño (manta térmica y aire acondicionado apagado). Se monitorizó la temperatura, la saturación arterial de oxígeno (SaO2), la frecuencia cardíaca y el ritmo cardíaco (ECG) y la tensión arterial de manera no invasiva. Se realizó la inducción anestésica con midazolán (0,01 mg/kg), fentanyl (5 µg/kg), atracurio como relajante muscular no despolarizante (0,6 mg/kg) y se realizó la intubación nasotraqueal con laringoscopio y espátula curva de Macintosh, colocando tubo convencional de 4,5 mm de diámetro interno, mientras se ventilaba previamente con máscara suministrando oxígeno al 100 %.
Este paciente afecto de artrogriposis distal no presentó signos de intubación difícil ni dificultad en la apertura de la boca. Se procedió a la canalización de la arteria radial y se colocó un catéter venoso central para medir la presión venosa central (PVC) y favorecer la administración de medicamentos. El paciente no presentó crisis de hipoxia durante la inducción anestésica, lo que permitió un mejor estado metabólico antes de comenzar la circulación extracorpórea (CEC).
Se mantuvo el nivel anestésico necesario con morfínicos sin empleo de agentes volátiles para prevenir episodios de hipertermia maligna, para lo cual se empleó fentanil a razón de 5-10 µg/kg), cada 1hora.
Este paciente tuvo en una ocasión una cifra elevada de glucemia preoperatoria que posteriormente resultó normal y no hubo antecedentes familiares ni elementos clínicos de una diabetes mellitus del lactante. No tuvo hipoglucemia durante el transoperatorio y la hiperglucemia transitoria estuvo relacionada con la administración de dextrosa al 5 % en la que se diluyen casi todos los medicamentos, además de la respuesta lógica al estrés quirúrgico que se desencadena durante la cirugía.
Se realizó la corrección del defecto cardíaco con un tiempo de CEC de 174 min y de pinzamiento aórtico de 73 min. Durante la extracorpórea se enfrió al paciente hasta que la temperatura rectal fue de 25 °C (hipotermia moderada). Se dieron tres dosis de cardioplejía cristaloide fría por la raíz aórtica para proteger el corazón. No hubo dificultad técnica en cerrar la comunicación interventricular, ampliar la salida del ventrículo derecho y luego abrir el anillo pulmonar. Se realizó un ecocardiograma transesofágico que mostró adecuada corrección quirúrgica. Se administró ácido épsilon-amino-caproico durante toda la intervención como profilaxis del sangramiento y se iniciaron infusiones continuas de inotrópicos que permitieron concluir la CEC: epinefrina (0,14 µg/[kg·min]) y dopamina (10 µg/[kg·min]) así como hemofiltración para alcanzar hematocrito de 35 y se traslada para la Unidad de Cuidados Intensivos (UCI) luego de mostrar estabilidad hemodinámica, ritmo sinusal y ausencia de sangramiento.
En su primer día de posoperatorio se manifiesta hiposaturación de 80% y se muestra hemodinámicamente inestable, dependiente de aporte de volumen y aumento de drogas adrenérgicas, taquicárdico (180 rpm), con diuresis menor de 1 mL/(kg·h). Presentó graves hiperglucemia (24 mmol/L) e hiperpotasemia (9 mmol/L), y esta última resultó difícil de controlar con todos los tratamientos empleados (incluyendo diálisis peritoneal). Presentó una primera parada cardíaca en fibrilación ventricular; se desfibriló con 30 J y se logró la asistolia. Se comenzó masaje cardíaco externo y se recuperó, pero necesitó noradrenalina y metaraminol debido a que presentó una vasoplejía. Se perpetuaron las manifestaciones clínicas del bajo gasto cardíaco con hipoxemia, hiperlactatemia progresiva (> 10 mmol/L) y acidosis metabólica, y presentó entonces una hipoglucemia (<1 mmol/L) que no respondía a la administración múltiple de dextrosa hipertónica. Ocurre una segunda parada cardíaca en asistolia sin respuesta a la epinefrina como fármaco de elección, empleada en bolos de 0,01 mg/kg cada 3 min; se mantuvo el masaje cardíaco externo continuo durante 40 min, sin mejores resultados. Este paciente tuvo un posoperatorio tórpido y falleció apenas 36 h después de la intervención quirúrgica. No se encontraron en la necropsia otras malformaciones.
DISCUSIÓN
Teniendo en cuenta los estudios epidemiológicos que reporta la literatura médica, la tetralogía de Fallot se asocia a anomalías extracardíacas en un 32,2 % de los casos, mientras que las anomalías cromosómicas se manifiestan en un 11,9 % de estos. Los síndromes cromosómicos que con más frecuencia se diagnostican en esta entidad son la trisomía del cromosoma 21 (síndrome de Down), del cromosoma 13 (síndrome de Patau) y del cromosoma 18 (síndrome de Edwards).2-4 Recientes avances en citogenética y técnicas de genética molecular han permitido la identificación de otros síndromes debido incluso a microdeleciones.
El presente caso ilustra una entidad clínico-genética poco frecuente en pediatría, como es el caso de un paciente con artrogriposis y cardiopatía congénita, producto de un parto gemelar. Se recogió el antecedente de hipertensión arterial por parte de la madre. El otro gemelo nació totalmente normal.
Ante este gemelo discordante para ambos defectos congénitos, se debe pensar en las causas extrínsecas fetales que los producen, tales como los eventos disruptivos vasculares que pueden ser generadores de ambos defectos. Las causas las extrínsecas son asimétricas, tienen pliegues de flexión exagerados con piel redundante en las articulaciones, y tienen las orejas grandes, elementos que no estaban presentes en este caso.
La presencia de otros signos clínicos orientadores como la simetría, la ausencia de pliegues de flexión, y la presencia de hoyuelos periarticulares, además del surco en la barbilla que recordaba la barbilla en H, hizo que no se descartara una artrogriposis distal de tipo II, por lo que se propuso manejo anestésico especial, debido a que se reportan complicaciones respiratorias, dificultad en la intubación (laringomalacia) y contracturas musculares con el empleo de halotano durante la anestesia,5 por lo cual no se utilizó para el mantenimiento de esta.
Desde el punto de vista cardiovascular, el paciente presentaba levocardia y situs solitus con concordancia de la conexión atrio-ventricular y ventrículo-arterial. El situs inversus puede encontrarse en menos de un 5 % de los pacientes mientras que la dextrocardia y el isomerismo atrial izquierdo son raros.6 La desviación anterosuperior del septum infundibular y su mal alineamiento son la esencia de los pacientes con esta cardiopatía. Este mal alineamiento ocurre entre el septum infundibular y el septum trabecular, y contribuye a la estenosis infundibular pulmonar y el defecto de septación ventricular típico de estos pacientes, lo cual se comprobó quirúrgicamente.6.
Kirklin y Barratt-Boyes han señalado que el cabalgamiento aórtico típico de estos pacientes está asociado a un grado variable de rotación de la raíz aórtica lo que contribuye además a la hipoplasia del tracto de salida del ventrículo derecho que se desarrolla en esta cardiopatía.7
La anatomía de los vasos coronarios, de gran importancia quirúrgica al realizar la ventrículotomía, fue normal en este paciente.
Teniendo en cuenta la fisiopatología, la tetralogía de Fallot diagnosticada en este paciente refleja la gravedad de la estenosis del tracto de salida del ventrículo derecho en presencia de una amplia comunicación interventricular, por lo que las presiones en los ventrículos y en la aorta eran las mismas, y el gasto pulmonar (Qp) y el gasto sistémico (Qs) dependieron de las resistencias respectivas al flujo. Presentaba además una importante hipertrofia ventricular derecha que limitaba el cuerpo de dicho ventrículo y empeoró el pronóstico.8-10
Se concluye que la artrogriposis constituye un síndrome con múltiples defectos congénitos de etiología compleja, que requiere de la interconsulta con el genetista clínico para establecer un diagnóstico de certeza, y puede asociarse a cardiopatías congénitas que complican el tratamiento y pronóstico de estos niños.
REFERENCIAS BIBLIOGRÁFICAS
1. Kalliainen LK, Drake DB, Edgerton MT, Grzeskiewicz JL, Morgan RF. Surgical management of the hand in Freeman-Sheldon syndrome. Ann Plast Surg 2003;50:45662.
2. Jones KL. Smith's Recognizable Patterns of Human Malformation. 6th Ed Montreal: W.S Saunders Company, 2006.
3. Stevenson RE, May JG, Goodman RM. Human malformations and related anomalies. Oxford: Oxford University Press. 1993.
4. Lantigua A. Introducción a la Genética Médica. 1 ed. La Habana:ECIMED; 2004.
5. Krakowiak PA, Bohnsack JF, Carey JC, Bamshad M. Clinical analysis of a variant of Freeman-Sheldon syndrome (DA2B). Am J Med Genet 1998; 76:938.
6. Van Praagh R, Van Praagh S, Nebesor R A. Tetralogy of Fallot: Underdevelopment of the pulmonary infundibulum and its sequelae. Am J Cardiol 1970;26: 25-33.
7. Kirklin J W, Barratt-Boyes B G. Cardiac Surgery, 2d ed. New York: Churchill Livingstone; 1993. Pp.861-1012.
8. Gatzoulis MA, Shore D, Yacoub M. Complete atrioventricular septal defect with tetralogy of Fallot: Diagnosis and management. Br Heart J 1994; 71:579-83.
9. Vricella LA, Gott VL, Cameron DE. Milestones in congenital cardiac surgery. En: Yuh DD, Vricella LA, Braumgartner WA. Manual of cardiothoracic surgery. Baltimore, Maryland: Mc Graw-Hill; 2007. Pp.989-98.
10. Haddadin A, Faraday N. Postoperative management of the cardiac surgical patient. En: Yuh DD, Vricella LA, Braumgartner WA. Manual of cardiothoracic surgery. Baltimore, Maryland: Mc Graw-Hill; 2007. Pp. 397-409.
Recibido: 26 de febrero de 2009.
Aprobado: 16 de abril de 2009.
Jacqueline Barrial Moreno. San Francisco 10112, Altahabana, Ciudad de La Habana, Cuba.
Correo electrónico: jaquibarrial@infomed.sld.cu

