










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión impresa ISSN 0034-7531
Rev Cubana Pediatr vol.83 no.4 Ciudad de la Habana oct.-dic. 2011
PRESENTACIÓN DE CASO
Proteinosis alveolar pulmonar
Pulmonary alveolar proteinosis
Concepción Sánchez Infante,I Anixia Serrano Sánchez,II Roberto Razón Behar,III Cristina Reyes LópezIV
IEspecialista de I Grado en Pediatría y Medicina General Integral. Diplomada en Terapia Intensiva Pediátrica. Hospital Pediátrico Universitario "William Soler". La Habana, Cuba.
IIEspecialista en I Grado en Anatomía Patológica. Hospital Pediátrico Universitario "William Soler". La Habana, Cuba.
IIIEspecialista de II Grado en Pediatría. Profesor Titular y Consultante. Hospital Pediátrico Universitario "William Soler". La Habana, Cuba.
IVEspecialista I Grado en Pediatría. Máster en Atención Integral al Niño. Hospital Pediátrico Universitario "William Soler". La Habana, Cuba.
RESUMEN
La proteinosis alveolar pulmonar es una enfermedad respiratoria crónica, caracterizada por alteración en el metabolismo del surfactante, lo que determina su acumulación anormal en el espacio alveolar. Es una enfermedad extremadamente rara. Se han reportado solamente 500 casos en la literatura. Se describió por primera vez en 1958. Se presenta un caso de proteinosis alveolar pulmonar en un lactante de 2 meses, con desnutrición proteico energética, que ingresa por dificultad respiratoria e hipoxemia, y, con imágenes radiológicas de tipo retículo-nodulillar, en vidrio deslustrado, en el cual se plantea inicialmente el diagnóstico de bronconeumonía. Ante la evolución desfavorable y no respuesta al tratamiento, se realizó un estudio para descartar enfermedades pulmonares crónicas. El paciente fallece y se confirma el diagnóstico por anatomía patológica. Se realiza una revisión del tema.
Palabras clave: proteinosis alveolar pulmonar, PAP, bronconeumonía.
ABSTRACT
The pulmonary alveolar proteinosis is a chronic respiratory disease characterized by surfactant metabolism alteration determining its abnormal accumulation in the alveolar space. It is a disease very rare and in literature only 500 cases have been reported; it was described for the first time in 1958. This is a case presentation of pulmonary alveolar proteinosis in an infant aged 2 months with energetic protein malnutrition admitted due to respiratory difficulty and hypoxemia and with radiologic images of the reticulonodulillary, in frosting glass, where initially is made the diagnosis of bronchopneumonia. In the face of unfavorable evolution and no response to treatment, a study was conducted to rule out chronic pulmonary diseases. Patient died confirming the diagnosis according to the pathologic anatomy. A review on subject is carried out.
Key words: pulmonary alveolar proteinosis, PAP, bronchopneumonia.
INTRODUCCIÓN
La proteinosis alveolar pulmonar (PAP) es una enfermedad extremadamente rara. Tiene una distribución mundial con una incidencia estimada de 0,36 casos por millón de habitantes.1 Se han reportado solamente 500 casos en la literatura.2,3 Fue descrita por primera vez por Rosen, Castleman y Liebow en 1958.4
Es una causa de enfermedad intersticial crónica pulmonar en los niños.5 En relación con su fisiopatología, se han realizado enormes progresos en los últimos años. Se describe como característica, el acúmulo de una cantidad excesiva y anormal de material proteináceo rico en lípidos y derivado del surfactante en los espacios aéreos alveolares, y en ocasiones, a nivel de la vías aéreas periféricas. Generalmente no induce inflamación, y muy raramente evoluciona hacia la fibrosis pulmonar.4,6
Clínicamente se caracteriza por disnea progresiva y tos, y evoluciona rápidamente hacia una insuficiencia respiratoria. Radiológicamente se manifiesta por la presencia de infiltrados bilaterales que recuerdan el edema pulmonar, e histológicamente por la acumulación y llenado alveolar por un material lipoproteináceo que es positivo a la coloración de ácido periódico-Schiff (examen de anatomía patológica PAS positivo).5
CASO CLÍNICO
Lactante masculino, procedente de área rural, nacido por cesárea (eclampsia y oligoanuria) a término, con un peso al nacer de 2 650 g, Apgar 9/9, BCG al nacer, lactancia materna exclusiva, padres sanos, y un hermano materno sano.
Asintomático, y con buena ganancia de peso hasta el mes de nacido, a los 45 días ingresa por un síndrome febril sin localización, lleva tratamiento antibiótico 1 semana y egresa. A los 2 meses en consulta de puericultura presenta palidez y polipnea, por lo que se ingresa en la Unidad de Terapia Intensiva Polivalente (UTIP) de su provincia, con el diagnóstico de bronconeumonía bacteriana y sepsis. Se mantiene en el hospital durante 33 días con estancia en UTIP en 3 ocasiones. Se observa discreta mejoría. Los diagnósticos planteados fueron: bronconeumonía por clínica y radiología, desnutrición proteico energética en la línea del marasmo, y anemia. En todas las radiografías de tórax se informaron lesiones inflamatorias en ambos campos pulmonares. Recibió tratamiento con diferentes antibióticos, además de transfusión de glóbulos y apoyo inmunológico. Se le realiza tomografía axial computarizada de tórax, que informa lesiones retículos nodulares de aspecto fibromatoso que recuerda imagen de carcinomatosis pulmonar. Se decide su traslado a nuestro centro hospitalario para completar el estudio.
Ingresa en el Hospital "William Soler", en el servicio de enfermedades respiratorias a los 3 meses y medio de nacido, con un peso de 3 800 g, afebril, con la piel y las mucosas hipocoloreadas, panículo adiposo disminuido, polipnea superficial, y una frecuencia respiratoria de 56 min, retracciones subcostales e intercostales. A la auscultación se comprueba buena ventilación de ambos campos pulmonares con escasos ruidos trasmitidos. El examen cardiovascular fue normal.
Exámenes de laboratorio al ingreso:
- Hemoglobina: 9,3 g/L
- Leucocitos: 23,9 x 109/L
- Polimorfonucleares: 74 %
- Linfocitos: 23 %
- Eosinófilos: 0,3 %
- Lámina periférica: plaquetas adecuadas, hipocromía anisocitosis. Leucocitos ligeramente aumentados, predominio polimorfonucleares.
- Conteo de reticulocitos: 5 %
- Plaquetas: 360 x 109/L
- Eritosedimentación: 2 mm/h
- Coagulograma: tiempo de sangrado prolongado que en exámenes seriados se normalizó.
- Química sanguínea (glicemia, transaminasas, creatinina, proteínas totales, colesterol, triglicéridos, fósforo, calcio, amilasa, amoniaco, fosfatasa alcalina, creatin fosfoquinasa [CPK] y ácido láctico): normales, y la albúmina, disminuida ligeramente.
- Examen parcial de orina: normal.
- En los gases arteriales se constató hipoxemia con pH normal. Hemocultivo negativo.
Examen radiológico: rayos X de tórax, imagen de tipo reticular en ambos campos pulmonares, confluente hacia los hilios, más acentuado en el hemitórax derecho. Imagen de pequeña atelectasia en hemitórax derecho. Área cardiovascular normal, y mediastino sin alteraciones.
US de tórax: no se ven lesiones parenquimatosas consolidantes, ni imagen de tumoración. Hay signos de hiperventilación pulmonar. El US abdominal arroja hígado que rebasa 1,5 cm el reborde costal, y el resto, normal.
Con todos los datos anteriores, se discutió como una enfermedad pulmonar intersticial pediátrica, y se inició estudio para descartar las múltiples entidades que la producen.
Microbiología: en exudado nasal y faríngeo de secreciones bajas, la flora fue normal. La IgM-IgG para citomegalovirus (CMV), virus Ebstein Barr (EBV), y virus del herpes simple (HSV) resultaron negativos, así como la proteína C reactiva (PCR) en orina para CMV, EBV y HSV. Igualmente, HIV y VDRL fueron negativos, como también ocurrió con la prueba de Mantoux, los cultivos bacteriológicos, micológicos y de bacilo de Koch.
La PCR en sangre fue positiva, el factor reumatoideo negativo, la IGA-2 disminuida, IGG, IGM resultaron normales, y los inmunocomplejos circulantes positivos. La alfa-feto-proteína resultó normal, la LDH aumentada, y los electrolitos en el sudor fueron normales.
El examen de rayos X de esófago, estómago y duodeno bajo pantalla no presentó compresiones esofágicas, ni reflujo gastroesofágico. El US de timo arrojó una hipoplasia tímica, el ecocardiograma no mostró malformaciones, ni hipertensión pulmonar. En el medulograma se descartaron enfermedades mieloproliferativas e infliltrativas. El estudio genético al bebé y a los padres para déficit de alfa 1 antitripsina fue negativo. Se realizó TAC de tórax de alta resolución cuyos resultados se presentan en la figura 1. No se pudo realizar lavado broncoalveolar, ni biopsia pulmonar por las condiciones clínicas del niño.
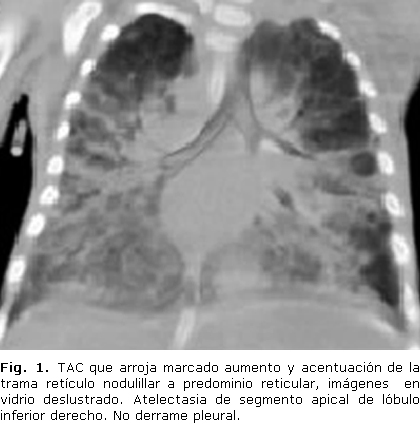
Durante su estadía hospitalaria el paciente presentó cuadros de hipoxemia, importante cianosis con livedu reticularis y aumento del trabajo respiratorio, por lo que necesitó traslado a Unidad de Cuidados Intensivos en varias ocasiones. Presentó fallo respiratorio progresivo, incremento de los requerimientos de oxígeno, y además, deterioro del estado general, pérdida de peso y emaciación, a pesar del adecuado aporte nutricional e inmunológico. El rayos xde tórax demostró aumento de opacidades pulmonares. Recibió tratamientos antibióticos con piperacilina con tazobactam, ciprofloxacino, trimetropim-cotrimoxazol (por la posibilidad de infección por Pneumocystis jiroveci), fluconazol (por lesiones micóticas orales), meronem, teicoplanina, ceftazidima, esteroides, factor de transferencia, albúmina y glóbulos rojos.
Precisó de ventilación mecánica a los 4 meses y medio. Se tomó muestra de secreciones por aspiración bronquial, que fue negativa de malignidad, ni de macrófagos cargados con hemosiderina. Fallece a los 5 meses por fallo respiratorio y disfunción múltiple de órganos.
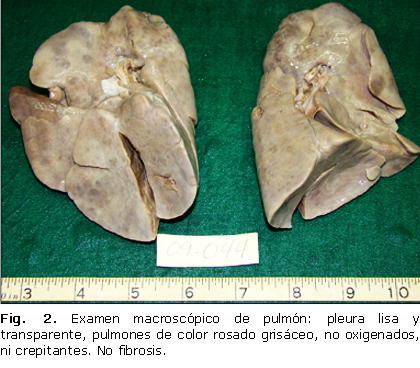
En la figura 2 se expone un examen macroscópico de pulmón. Al corte, se observa superficie de aspecto carnoso, de color rojo carmelita y seco. No fibrosis. Tráquea y bronquios con material espeso rosado. En el examen histológico de pulmón se comprobaron alveolos totalmente ocupados por material rosado granuloso y PAS positivo, así como la presencia de escasas células de inclusión citomegálica. Se concluye: PAP y enfermedad de inclusión citomegálica.
DISCUSIÓN
Actualmente la PAP es enfocada como un síndrome basado en la comprensión de los mecanismos de la enfermedad en:
Síndrome de disfunción del surfactante (PAP congénita): puede ser resultado de una rara mutación genética, se debe a un déficit congénito de la proteína B y C del surfactante (SP-B) (SP-C), lo que da como resultado un surfactante disfuncional, o bien se genera a partir de la mutación de la cadena Bc del receptor del factor estimulador de colonias de granulocitos y macrófagos (GM-CSF).7-9 Recientemente se han descrito mutaciones de un gen localizado en el brazo corto del cromosoma 16, gen que codifica para la ATP-binding cassette protein A3 (ABCA3).10 Las proteínas del surfactante pueden ser hidrofílicas, como la SP-A y SP-D, o hidrofóbicas como la SP-B y SP-C. Las primeras son esenciales para la defensa inmunológica y la modulación de la respuesta alérgica del pulmón, mientras las hidrofóbicas reaccionan con los lípidos del surfactante para facilitar su absorción a la interfase líquido-gaseosa.11
La proteína SP-B se codifica en el brazo corto del cromosoma 2 y cuenta con 11 axones. El rol de la SP-B probablemente tenga relación con el empaquetamiento de los fosfolípidos en los cuerpos lamelares, y es esencial en la proteólisis de proSP-C. La deficiencia de SP-B fue la primera causa genética conocida del síndrome de distrés respiratorio (SDR), y la mutación del codón 121 es la más frecuente, aunque suele ser única para cada familia afectada. La sintomatología aparece en las primeras 48 h de vida, y las técnicas de imagen muestran un patrón en vidrio deslustrado, como una enfermedad por membrana hialina. Microscópicamente muestra una proteinosis alveolar con un material eosinófilo PAS-diastasa positivo en los alveolos -posiblemente proSP-C aberrantemente procesada- y descamación de neumocitos con abundantes macrófagos. El paciente fallece por insuficiencia respiratoria a los 3-6 meses sin que se obtenga respuesta con SP-B exógena.12
El cuadro clínico se caracteriza por dificultad respiratoria progresiva y severa, en niños a término, con resistencia a tratamiento habitual por neumopatía, que evolucionan a falla respiratoria, y cuya única posibilidad de sobrevida lo constituye el trasplante pulmonar.13
Proteinosis alveolar primaria o idiopática del adulto: es rara en niños, representa más del 90 % de los casos de esta entidad, ocurre en sujetos previamente sanos, sin antecedentes hereditarios, ni de exposición ambiental u ocupacional.14 Es una manifestación de un fenómeno de autoinmunidad con producción de anticuerpos antifactor estimulador de colonias para granulocitos y macrófagos (GM-CSF). Se produce una alteración funcional o una reducción en el número de los macrófagos alveolares, y esto conduce a una disminución del aclaramiento del surfactante, y por tanto, a la acumulación de material proteico en los alvéolos.15,16
Proteinosis alveolar secundaria: representa entre el 5-10 % de los casos con esta enfermedad.14 Aparece asociada a otras que provocan una alteración cuantitativa o cualitativa de los macrófagos alveolares. Se ha asociado a enfermedades malignas (leucemia mieloide aguda y crónica, leucemia linfoide aguda y crónica, mieloma múltiple, mielodisplasia), a inmunodeficiencias (Sida, inmunodeficiencias combinadas, amiloidosis, hipogammaglobulinemia, púrpura trombocitopénica idiopática, anemia aplástica), a infecciones crónicas (citomegalovirus, Micobacterium tuberculosis, Pneumocistis jiroveci, histoplasmosis), a la exposición de polvos inorgánicos (sílice, aluminio, cemento, dióxido de titanio, dióxido de nitrógeno), y a la inhalación de humos tóxicos (cenizas volcánicas).17-19
Nuestro paciente reunía los elementos clínicos, radiológicos y anátomo-histológicos de la PAP congénita, porque desde las 6 sem de vida aparecieron signos de dificultad respiratoria, requerimientos de oxígeno, que siguieron un curso progresivo con deterioro del estado general y detención de su curva ponderal, fallece con menos de 6 meses por insuficiencia respiratoria. Presentó, igualmente, elevación del ácido láctico deshidrogenasa (LDH) en plasma, que es secundario a la hipoxia, ayuda al diagnóstico, pero no es patognomónico de esta entidad. Otros autores han hecho referencia a la elevación de la LDH en pacientes con PAP, relacionándolo con la severidad de la enfermedad.6,20,21 También encontramos hipoxemia con pH normal, lo cual coincide con la literatura.2,6,20
La infección por citomegalovirus informada en el estudio histopatológico post mortem, se relaciona con una infección intrahospitalaria, pues los exámenes iniciales para esta infección y otras, tanto en suero como orina, fueron negativos; además, tenía factores de riesgo para infección nosocomial, como son: la edad, la larga estadía hospitalaria, y la desnutrición proteica energética, a la que se añade hipoplasia tímica y enfermedad intersticial crónica pulmonar. Un rasgo importante de la PAP es la susceptibilidad que presenta para infecciones pulmonares, incluyendo a gérmenes oportunistas, por lo que los procesos infecciosos son las principales complicaciones que se presentan.8,13
La radiografía simple de tórax puede ser indistinguible de otras enfermedades intersticiales respiratorias crónicas. El patrón radiológico consiste en un infiltrado fino y difuso que se irradia de los hilios hacia la periferia en forma de "alas de mariposa", las lesiones son bilaterales y simétricas; a veces este infiltrado toma aspecto reticulonodular diseminado en "parches", con áreas de confluencia asimétricas, de distribución central o periférica, sin la presencia de cardiomegalia, ni derrame pleural, y sin compromiso de los ángulos cardiofrénicos.2,5,22 En la tomografía axial computarizada de tórax se describen desde opacidades nodulares mal definidas, a grandes áreas de opacidades de vidrio deslustrado bilateral difuso, con distribución hiliar y perihiliar, engrosamiento de septos interlobulillares, distribución geográfica o en parches, con zonas de parénquima indemne. La combinación de imágenes nos da el patrón característico de proteinosis conocido como "pavimentación".2,8 El caso descrito por nosotros en el rayos x de tórax tenía imagen de tipo reticular en ambos campos pulmonares, confluente hacia los hilios. En radiografías de tórax evolutivas, se observó incremento de las opacidades pulmonares de forma difusa. En la TAC se describieron imágenes en vidrio deslustrado.
El diagnóstico de la PAP se confirma mediante biopsia pulmonar a "cielo abierto", o por videotoracoscopia; también mediante el lavado broncoalveolar, al demostrar el aspecto clásico del líquido recuperado y su estudio citoquímico (líquido estéril, opaco, de color crema o beige, que sedimenta rápidamente formando una capa sólida insoluble y un líquido claro), y además, la presencia de numerosos macrófagos alveolares con gran cantidad de proteínas y lípidos en su interior (tinciones PAS y Sudán).3,5,8,23 Fue imposible realizar lavado broncoalveolar y biopsia pulmonar dada la gravedad del paciente.
Otra forma de confirmar el diagnóstico desarrollada en los últimos años es la cuantificación de anticuerpos, antifactor estimulador de colonias para granulocitos y macrófagos.2,6,21
Para la PAP congénita no existe tratamiento efectivo. Solo se indican medidas de soporte con oxígeno, ventilación asistida u oxigenación de membrana extracorpórea. La mayoría de los pacientes afectados fallecen en los primeros 6 meses de vida. Se ha considerado el trasplante pulmonar doble como una opción terapéutica alternativa.5,24
En la PAP secundaria, el tratamiento será de la enfermedad que la ocasione o el factor de exposición identificado. En la forma idiopática, en cambio, es necesario el lavado broncopulmonar total25 y la terapia con factor estimulante de colonias para macrófagos/granulocitos.26,27 Con la utilización del lavado broncoalveolar, el pronóstico ha mejorado, sobre todo, en adultos. Se observan mejorías espectaculares transitorias con aclaramiento radiológico y mejoría del intercambio gaseoso. Durante las recaídas es necesario repetir los lavados. Los niños en general tienen poca respuesta con la utilización de este proceder.5 Si se sospecha infección sobreañadida, se debe tratar agresivamente con antibióticos.
REFERENCIAS BIBLIOGRÁFICAS
1. Ben-Dov I, Kishinevski Y, Roznman J, Soliman A, Bish-ara H, Zelligson E, et al. Pulmonar Alveolar Proteinosis in Israel. Isr Med Assoc J. 1998;1:75-8.
2. Huizar, Isham, Kavuru, Mani S. Alveolar proteinosis syndrome: pathogenesis, diagnosis, and management. Curr Opin Pulm. Med 2009;15(5):491-8.
3. Seymour JF, Presneill JJ. Pulmonary alveolar proteinosis progress in the first 44 years. Am J Respir Crit Care Med. 2002;166:215-35.
4. Rosen SH, Castleman B, Liebow AA. Pulmonary alveolar proteinosis. N Engl J Med. 1958;258:1123-42.
5. Razón BR. Otras enfermedades que afectan principalmente los pulmones. Proteinosis alveolar pulmonar. En: Autores cubanos. Pediatría III. La Habana: Editorial Ciencias Médicas; 2007. p. 1079-80.
6. Param ST, Flores SM, Bolbaran SJP, Díaz CA. Proteinosis Alveolar Pulmonar. Caso Clínico. Rev Chil Pediatr. 2007;78(2):176-82.
7. Tracey B, Russell D, Burgess S, Malur A, Mani S Kavuru, Thomassen MJ. Autoantibodies against granulocyte macrophage colony-stimulating factor are diagnostic for pulmonary alveolar proteinosis. Am J Respir Cell Mol Biol. 2002;27:481-6.
8. Mejía MH, Alonzo MD, Suarez LT, Gaxiola GM, Carrillo RJG. Proteinosis Alveolar Pulmonar. Neum y Cirug tórax. 2006;65(S3):S15-S23.
9. Martinez-Moczygemba M, Doan ML, Elidemir O. Pulmonary alveolar proteinosis caused by deletion of the GM-CSFRalpha gene in the X chromosome pseudoautosomal region 1. J Exp Med. 2008;205:2711-6.
10. Yokota T, Matsumura Y, Ban N. Heterozygous ABCA3 mutation associated with nonfatal evolution of respiratory distress. Eur J Pediatr. 2008;167:691-3.
11. Hartl D, Griese M. Interstitial lung disease in children-genetic background and associated phenotypes. Respiratory Research. 2005;36:32-48.
12. Bullard JE. ABCA3 mutations associated with pediatric interstitial lung diseases. Am J Crit Care Med. 2005;172:1026-31.
13. Trapnell BC, Witsett JA, Nakata K. Pulmonary Alveolar Proteinosis. N Engl J Med. 2003;349:2527-39.
14. Presneill J, Nakata K, Inove Y, Seymour JF. Pulmonary alveolar proteinosis. Clin Chest Med. 2004;25:593-613.
15. Lin FC, Chang GD, Chem MS. Chen YC, Chang SC. Clinical significance of anti-GM-CSF antibodies in idiopathic pulmonary alveolar proteinosis. Thorax. 2006;61:528-34.
16. Kitamura T, Tanaka N, Watanave J, Uchida, Kanegasaki S, Yamada Y, et al. Idioppathic pulmonary alveolar proteinosisas an autoinmune disease with neutralizing antibodyagainst Granulocyte/Macrofhage colony-stimulating factor. J Exp Med. 1999;190(6):875-80.
17. Buechner H, Ansari A. Acute silicoproteinosis: a new pathologic variant of acute silicosis insand-blasters, characterized by histologic features resembling alveolar proteínosis. Dis Chest. 1969;55:274-8.
18. Ruben F, Talamo T. Secondary pulmonary alveolar proteínosis occurring in two patients with acquired immune deficiency syndrome. Am J Med. 1986;80:1187-90.
19. Doyle A, Balcerzac S, Wells C, Crittenden J. Pulmonary alveolar proteinosis with hematologic disorders. Arch Intern Med. 1963;112:940-6.
20. Latorre JJ, Liberman RL, Campos PZ, Ariztia AA. Proteinosis Alveolar Pulmonar en un lactante. Rev Chil de Pediatría. 1973;44(2):147-50.
21. Seymour JF, Doyle IR, Makata K. Relationship of anti GM-CSF Antibody concentration surfactant protein A and B levels, and serum LDH to pulmonary parameters and response to GM-CSF therapy in patients with idiopathic alveolar Proteinosis. Thorax. 2003;58:252-7.
22. Lee KN, Levin DL, Webb WR, Chen D, Storto ML, Golden JA. Pulmonary alveolar proteinosis: high-resolution CT, chest radiographic, and functional correlations. Chest. 1997;111:989-95.
23. Burkahalter A, Silverman JF, Hopkins MB 3rd, Gresinger KR: Bronchoalveolar lavage cytology in pulmonary alveolar proteinosis. Am J Clin Pathol. 1996;106:504-10.
24. Mahut B, Delacourt C, Scheinmann P. Pulmonary alveolar proteinosis: experience with eight pediatric cases and a review. Pediatrics. 1996;1:117-22.
25. Ben-Abraham R, Greenfeld A, Rozenman J. Pulmonary alveolar proteinosis: step-by-step perioperative care of whole lung lavage procedure. Heart Lung. 2002;31:43-9.
26. Yamamoto H, Yamaguchi E, Agata H. A combination therapy of whole lung lavage and GM-CSF inhalation in pulmonary alveolar proteinosis. Pediatr Pulmonol. 2008;43:828-30.
27. Seymour JF,Presneill JJ,Schoch OD, Downie GH, Moore PE, Doyle IR. Therapeutic efficacy of granulocyte-macrophage colony-stimulating factor in patients with idiopathic acquired alveolar proteínosis. Am J Respir Crit Care Med. 2001;163:524-31.
Recibido: 19 de mayo de 2011.
Aprobado: 27 de junio de 2011.
Concepción Sánchez Infante. Hospital Pediátrico Universitario "William Soler". Avenida San Francisco # 10 112, reparto Altahabana, municipio Boyeros. La Habana, Cuba. Correo electrónico: csifante@infomed.sld.cu

