










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión impresa ISSN 0034-7531
Rev Cubana Pediatr vol.85 no.2 Ciudad de la Habana abr.-jun. 2013
PRESENTACIÓN DE CASO
Primer caso registrado en Cuba de nefropatía C1q
First case of C1q nephropathy in Cuba
MSc. Dra. Neri Georgina Campañá Cobas,I Dr. Agustín Chong López,II Dr. Sandalio Durán Álvarez,I Dr. Severino Hernández Hernández,I MSc. Dr. Mario Valdés MesaI
IHospital "William Soler". La Habana, Cuba.
IIHospital "Hermanos Ameijeiras". La Habana, Cuba.
RESUMEN
La nefropatía C1q es una glomerulopatía no comprendida completamente y con algunas controversias conceptuales, pero con característica inmunológica distintiva (depósito dominante o co-dominante de C1q) y no evidencia clínica ni serológica de lupus eritematoso sistémico. Se presenta paciente masculino que comienza a los 10 meses de edad con un síndrome nefrótico con hematuria, hipertensión arterial e insuficiencia renal. Se realiza una primera biopsia renal y se plantea una esclerosis mesangial difusa, pero su evolución posterior con respuesta parcial a la prednisona y el mantenimiento de proteinuria en rango nefrótico con normalización de los parámetros humorales, nos lleva a realizar una segunda biopsia renal que arroja, por la inmunofluorescencia, una nefropatía C1q.
Palabras clave: síndrome nefrótico, nefropatía C1q, hematuria, proteinuria.
ABSTRACT
C1q nephropathy is a poorly understood glomerulopathy with some conceptual controversies, but with a distinctive immunologic characteristic (dominant or co-dominant deposit of C1q) and neither clinical nor serological evidence of systemic erythematous lupus. This is the case of a male patient who began suffering nephritic syndrome with hematuria, blood hypertension and renal failure at 10 months of age. A first renal biopsy was performed to detect diffuse mesangeal sclerosis; however after partial response of the patient to prednisone therapy and to maintenance treatment of proteinuria in nephritic range, with normalization of humoral parameters, then a second renal biopsy was performed with immunofluorescence. The final result was C1q nephropathy.
Key words: nephrotic syndrome, C1q nephropathy, hematuria, proteinuria.
INTRODUCCIÓN
La nefropatía C1q (NC1q) es una controversial glomerulopatía, descrita por primera vez por Jennette y Hipp en 1985,1 como una forma de síndrome nefrótico idiopático causada por depósito dominante o co-dominante mesangial de C1q, de al menos 2+ o más de intensidad de una escala de 0 a 4+, y no evidencia clínica ni serológica de lupus eritematoso sistémico. Clínicamente la mayoría de los pacientes se presentan con síndrome nefrótico córtico resistente o proteinuria asintomática resistente a los esteroides, y algunos con insuficiencia renal.2,3
Después de esta primera descripción se han reportado varios estudios en los que se ha demostrado que esta entidad puede presentarse a cualquier edad, con distintas formas clínicas de presentación y variadas alteraciones histológicas.4-9
La inmunofluorescencia muestra depósitos de C1q de al menos 2+ o más de intensidad, además depósito de IgG (90 %), IgM (94 %), C3 (90 %) y la IgA es menos común.1-3 En la microscopía electrónica alrededor del 90 % de los casos revela depósitos electrodensos subyacentes de la membrana basal glomerular, y en casos raros depósitos subendoteliales y subepiteliales.2,3
En cuanto a la conducta, muchos pacientes son tratados con esteroides, también se ha utilizado tratamiento con inmunosupresores (ciclofosfamida, ciclosporina, micofenolato mofetil y tracolimus) e inhibidores de la encima de conversión de angiotensina.7,10,11 La evolución de la función renal, a largo plazo, en la nefropatía C1q no está clara; en pacientes con cambios de glomeruloesclerosis segmentaria focal (GESF) parece haber un curso más agresivo. La enfermedad es frecuentemente resistente a esteroides o hay recaídas.
Nuestro objetivo es la descripción de un niño con una entidad clínica histológica no reportada previamente en Cuba, en el que se comprueba, después de realizar dos biopsias renales, una nefropatía C1q por la inmunofluorescencia.
PRESENTACIÓN DEL CASO
Paciente masculino, blanco, con antecedentes de ultrasonido prenatal normal, parto eutócico a las 39,1 semanas, peso al nacer de 2 400 g, sin consanguinidad paterna ni antecedentes familiares de nefropatía, que a la edad de 10 meses ingresa por un cuadro de edema generalizado y proteinuria. Se diagnostica un síndrome nefrótico, y comienza tratamiento con prednisona (60 mg/m2/día). A los 7 días comienza con hematuria macroscópica e hipertensión arterial, y es remitido a nuestro centro, a donde llega con edema generalizado (anasarca), hipertensión arterial (130/90) y hematuria macroscópica. En los exámenes complementarios presenta: proteinuria (5,4 g/24 h), hipoproteinemia (35 g/L), hipoalbuminemia (18 g/L), hipercolesterolemia (22 mmol/L), hipertrigliceridemia (10 mmol/L), anemia (79 g/L), creatinina (128 µmol/L), hipocalcemia (1,9 mmol/L), hiperfosfatemia (5,39 mmol/L), C3 y C4 normales, anticuerpos antinucleares, anti-DNA y anticuerpos anticitoplasma de neutrófilos (ANCA) negativos, ecografía renal con ambos riñones hiperecogénicos y aumentados de tamaño.
Se realiza una primera biopsia renal, que, por lo escaso de la muestra, nada más se realiza microscopía óptica (MO), que arroja 11 glomérulos que muestran proliferación mesangial discreta y global con esclerosis, existe ligera ectasia tubular focal que contiene material proteico tipo proteína de Tamn Horsfall, algunos con microcalcificaciones. Se observa reabsorción proteica en el epitelio tubular proximal, y ligera fibrosis asociada a escaso infiltrado inflamatorio intersticial, que plantea como diagnóstico un síndrome nefrótico infantil con discreta esclerosis mesangial difusa en estadio temprano (figura 1 A, B, C, D).
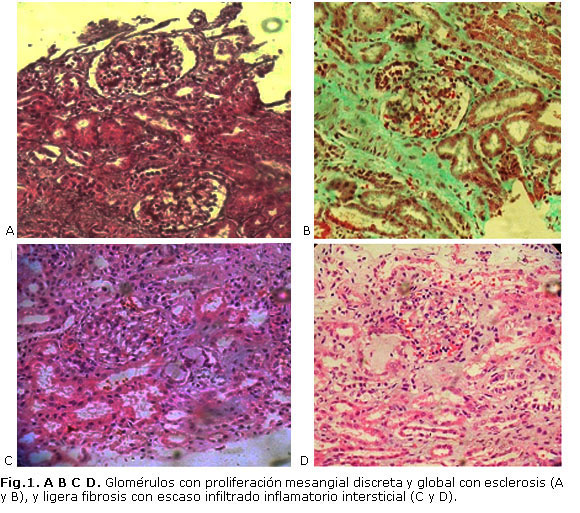
Con este resultado se suspende la prednisona a las 4 semanas y se inicia tratamiento con aporte alto de proteína, administración de albúmina y furosemida, por el estado de anasarca que presenta el paciente, así como sales de hierro, vitaminoterapia, carbonato de calcio y antibiótico profiláctico, por las infecciones recurrentes. A los 6 meses de evolución no es necesario administrar más albúmina ni furosemida, y humoralmente el paciente comienza a normalizar todos los complementarios, pero mantiene la proteinuria en rango nefrótico, con un desarrollo nutricional adecuado, por lo que se realiza una segunda biopsia renal con MO e inmunofluorescencia (IF), pero sin microscopía electrónica, y se reporta:
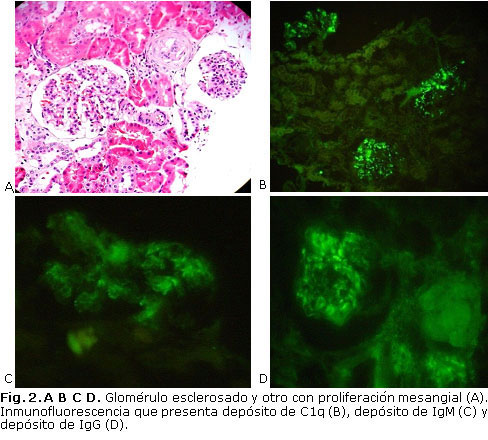
- MO: 21 glomérulos, 6 de ellos globalmente esclerosados, el resto con ligera proliferación mesangial segmentaria. Atrofia tubular relacionada con los glomerúlos esclerosados, y muy escaso infiltrado linfocitario intersticial. No hay fibrosis intersticial ni lesión vascular (figura 2 A).
- IF: depósitos segmentarios mesangiales y periféricos de IgG++, IgM++, C3+ y predominante C1q+++ (figura 2 B, C, D). Luego de la IF se concluye que es una nefropatía C1q.
DISCUSIÓN
La NC1q es una glomerulopatía no comprendida completamente y con algunas controversias conceptuales, pero con característica inmunológica distintiva (depósito dominante o co-dominante de C1q).10 Muchos casos de NC1q han sido descritos como nefritis lúpica "seronegativa" en pacientes con hallazgo histológico de nefritis lúpica, pero sin evidencia serológica o clínica de lupus eritematoso.12 Markowitz describe esta nefropatía como una variante de la GESF, y afirma que la NC1q cae dentro del marco cambio mínimo glomerular/GESF. Es poco frecuente, subdiagnosticada y aún no bien estudiada, debido a lo cual no existe consenso de considerarla como una entidad aislada, o bien incluirla dentro del espectro de la esclerosis segmentaria focal.10
Nuestro paciente tuvo una primera biopsia renal que plantea una esclerosis mesangial difusa temprana por las alteraciones presentadas al microscopio de luz, que conjuntamente con la clínica de síndrome nefrótico insuficiencia renal y el comienzo en el primer año de vida nos hizo pensar que se tratara de esta enfermedad, pero la evolución clínica y la normalización de las alteraciones humorales pusieron en duda este diagnóstico. Se realiza una segunda biopsia renal al año y medio de la primera, por la proteinuria en rango nefrótico que mantiene el paciente, que arroja una glomeruloesclerosis global y focal con proliferación mesangial segmentaria, diagnóstico descriptivo que se correlaciona con el dominio en la inmunofluorescencia de C1q.
En nuestro paciente no se puede demostrar que en el momento de la primera biopsia tuviera depósitos de C1q por no poder realizar inmunofluorescencia, pero se piensa que pudiera no haberla tenido, al igual que 3 pacientes con síndrome nefrótico en un estudio realizado por Vizjak y otros, que en una primera biopsia fueron C1q negativos, pero en la segunda fueron positivos, así como en un paciente que fue positiva en la primera y negativa en la segunda. Ellos estudiaron en un periodo de 20 años 72 pacientes, en los que el 68,1 % fueron masculinos y 28 en edad de 2 a 17 años. Al microscopio de luz 27 pacientes no tenían lesión, 11 presentaban GESF, 20 glomerulonefritis proliferativa y 14 con otros tipos de lesiones. El síndrome nefrótico se presentó en 17 de los que no presentaban lesiones y en los 11 con GESF.8
Levart y otros, en estudio retrospectivo de 10 años en 137 biopsias renales realizadas en 131 niños, encontraron 12 que presentaban nefropatía por C1q, de los cuales 8 tenían un síndrome nefrótico, 3 con proteína no nefrótica asociada a microhematuria, hipertensión arterial o insuficiencia renal, y 1 con proteinuria nefrótica que evolucionó a la insuficiencia renal terminal. El patrón histopatológico más frecuente fue la GESF, con o sin proliferación mesangial difusa, en 6 de los 12 casos.11
En estudio hecho por Iskandar y otros con 15 niños, 10 eran del sexo femenino y 11 blancos. Se presentaron con síndrome nefrótico 9, los cuales recibieron tratamiento con esteroides previo a la biopsia renal, de ellos 5 no respondieron, 2 fueron córtico dependientes y 2 tuvieron recaídas frecuentes; 4 pacientes tenían síndrome nefrítico y 2 con proteinuria persistente.13 En otro estudio realizado por Markowitz en 19 pacientes, el 73,7 % fueron afronorteamericanos y del sexo femenino, la proteinuria de rango nefrótico se presentó en el 78,9 % de los pacientes, y de estos, el 50 % tenía un síndrome nefrótico, la insuficiencia renal estuvo presente en el 27,8 % y la hematuria en el 22,2 %. La serología para lupus eritematoso sistémico y el virus de inmunodeficiencia humana (VIH) fue negativa, los niveles de complemento sérico fueron normales, y no hubo evidencia clínica ni serológica de enfermedad sistémica, autoinmune o infecciosa.10
En estudio realizado en Japón por Hisano y otros, en 61 pacientes, el 59 % se presentó con anomalía urinaria asintomática y el 41 % con síndrome nefrótico. Todos los pacientes con síndrome nefrótico fueron tratados con prednisona y/o ciclosporina, además de 9, en el grupo asintomático, que recibieron prednisona.14
En la descripción inicial de la nefropatía C1q realizada por Jennette al microscopio de luz, señala un amplio espectro de características patológicas desde anomalías glomerulares no significativas hasta GESF y glomerulonefritis proliferativa.1 En los 15 casos de Iskandar, 8 no presentaban anomalías histológicas, 7 presentaban GESF y 3 de estos con proliferación mesangial.13 Markowitz encuentra de las 19 biopsias renales, 17 con GESF, y de ellas 6 con variante colapsada, 2 con celular y 9 con variante no específica. La atrofia tubular o fibrosis intersticial de ligera a severa estuvo presente en 17 pacientes.5
Los pacientes con síndrome nefrótico particularmente con GESF tienen una pobre respuesta a los corticoesteroides, mientras que pacientes que presentan anomalías urinarias asintomáticas mantienen función renal normal. En el caso de nuestro paciente tuvo una respuesta parcial a los esteroides porque mejoró el cuadro humoral, pero mantiene la proteinuria en rango nefrótico, por lo que su pronóstico es incierto, lo que obliga a su seguimiento y control a largo plazo.
Agradecimientos
Nuestro agradecimiento a los doctores Jean Pierre Guignard y Marie Claire Gubler por la ayuda en el diagnóstico del paciente.
REFERENCIAS BIBLIOGRÁFICAS
1. Jenette JC, Hipp CG. C1q Nephropathy: a distinct pathology entity usually causing nephrotic Syndrome. Am J Kidney Dis. 1985;6:103-10.
2. Fogo AB. Minimal change disease and Focal Segmental Glomerulosclerosis. En: Fogo AB, Brujin JA, Cohen RB, Jenette JC. Fundamental of Renal Pathology. New York: Springer Science; 2006. p. 40-9.
3. Focal Segmental Glomerulosclerosis. En: Non-neoplastic. Kidney diseases. King DW (Ed). Washington, DC.: Advisoy Board; 2005. p. 125-59.
4. Nishida M, Kawakatsu H, Okumura Y, hamaoka K. C1q nephropathy with asymptomatic urine abnormalities. Pediatr Nephrol. 2005;20:1669-70.
5. Srivastova T, Chaldha V, Taboada EM, Alon US. C1q nephropathy presenting as rapidly progressive crescentica glomerulonephritis. Pediatr Nephrol. 2000;14:976-9.
6. Sardani Y, Qin K, Haas M, Aronson AJ, Rosenfield RL. Bartter syndrome complicated by immune complex nephropathy. Case report and literature review. Pediatr Nephrol. 2003;18:913-8.
7. Roberti I, Baqi N, Vyas S, Kim DU. A single center study of C1q nephropathy in children. Pediatr Nephrol. 2009;24:77-82.
8. Vizjak A, Ferluga D, Hvala A, Lindia J, Lervart TK, Jura V, et al. Pathology, presentations and outcomes of C1q Nephropaty. Am Soc Nephrol. 2008;11:2237-44.
9. Taggart L, Harris A, El-Dart S, Iorember F. C1q Nephropathy in a child presenting with recurrent gross hematuria. Pediatr Nephrol. 2010;25:165-8.
10. Maskowitz GS, Schwimmer JA, Barry Stokes M, Nasr S, Seigler RL, Valeri AM. C1q Nephropathy: a variant of focal segmental glomerulosclerosis. Kidney International. 2003;641:232-40.
11. Lervat KT, Kenda RB, Cavié MA, Ferluga D, Hvala A, Vizjak A. C1q Nephropathy in children. Pediatr Nephrol. 2005;20:1756-61.
12. Sharman A, Furness P, Feehally J. Distinguishing C1q nephropathy from lupus nephritis. Nephrol Dial Transplant. 2004;19:1420-6.
13. Iskandar SS, Browning MC, Lorentz WB. C1q Nephropathy: A Pediatric Clinicopathology Study. Am J Kidney Dis. 1991;18:459-65.
14. Hisano S, Fukuma Y, Segawa Y, Niimi K, Kaku Y, Hatoe K, el at. Clinicopathologic correlation and outcome of C1q nephropathy. J Am Soc Nephrol. 2008;6:1637-43.
Recibido: 3 de octubre de 2012.
Aprobado: 19 de octubre de 2012.
Neri Georgina Campañá Cobas. Hospital Pediátrico Universitario "William Soler". San Francisco # 10 112, Reparto Altahabana, municipio Boyeros. La Habana, Cuba. Correo electrónico: nerycc@infomed.sld.cu
