










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr vol.87 no.3 Ciudad de la Habana jul.-set. 2015
PRESENTACIÓN DE CASO
Detección de un caso síndrome de Pallister-Killian diagnosticado por citogenética convencional
Detection of Pallister-Killian syndrome case through conventional cytogenetic diagnosis
MSc. Carolina Isaza de Lourido,I Nathaly Duque-Moncaleano,II MSc. Felipe Ruíz-Botero,III Dr.C. Harry PachajoaIII
IUniversidad del Valle. Cali, Colombia.
IIEstudiante de Medicina. Universidad Icesi. Cali, Colombia.
IIIUniversidad Icesi. Cali, Colombia.
RESUMEN
El síndrome de Pallister-Killian es producido por una tetrasomía del brazo corto del cromosoma 12 en algunas células del cuerpo, debido a la presencia de un isocromosoma (12p), mientras que el resto de las células tienen un complemento cromosómico normal, fenómeno al cual se le denomina mosaicismo cromosómico. Se considera de ocurrencia esporádica, con muy bajo riesgo de recurrencia, y afecta igualmente a hombres y mujeres. El síndrome de Pallister-Killian o tetrasomía 12p en mosaico, tiene un fenotipo amplio y no específico, que se caracteriza con mayor frecuencia por hipotonía, retardo mental severo, sordera y convulsiones, que pueden empeorar con la edad. Se reporta el caso de un niño diagnosticado con síndrome de Pallister-Killian en Colombia, y este reporte hace referencia a las dificultades para realizar el diagnóstico de la anomalía cromosómica, ya que no se sospecha el síndrome y el cariotipo convencional puede dar resultado negativo.
Palabras clave: síndrome de Pallister-Killian, anomalía congénita, genética.
ABSTRACT
Pallister-Killian syndrome occurs from a tetrasomy of chromosome 12 short arm in some body cells due to the presence of isochromosome (12p) whereas the rest of cells have normal chromosomal complement. This phenomenon is called chromosomal mosaicism. It is considered to occur sporadically, with very low chance of recurrence and affects both women and men. Pallister-Killian syndrome or tetrasomy 12p mosaicism has wide non-specific phenotype characterized by higher frequency of hypotonia, severe mental retardation, deafness and seizures that may worsen as age increases. This is the report of a child diagnosed with Pallister-Killian syndrome in Colombia, which makes reference to difficulties in diagnosing a chromosomal anomaly, since this syndrome is not suspected and the testing for conventional karyotype may provide negative results.
Keywords: Pallister-Killian syndrome, congenital anomaly, genetics.
INTRODUCCIÓN
El síndrome de Pallister-Killian (SPK) (MIM: 601803) es causado por una alteración cromosómica estructural compleja, que deja como resultado una tetrasomía del brazo corto del cromosoma 12 en algunas células del cuerpo, debido a la presencia de un isocromosoma (12p), mientras que el resto de las células tienen un complemento cromosómico normal, lo que se denomina mosaicismo cromosómico.1 El mecanismo por el cual se produce esta alteración genética no es claro. Este síndrome se considera de ocurrencia esporádica, con muy bajo riesgo de recurrencia, y afecta a hombres y mujeres por igual.2 Fue descrito inicialmente de manera independiente por Pallister y otros en 1977, y Killian en 1983.3,4
El SPK o tetrasomía 12p en mosaico, tiene un fenotipo amplio, con hallazgos característicos distintivos, como es la ausencia de cabello en regiones froto-temporales en los primeros años de vida, filtrum largo con labio superior fino y arco de cupido muy marcado; y hallazgos no específicos, que se caracterizan, con mayor frecuencia, por hipotonía, retardo mental severo, sordera y convulsiones, que pueden empeorar con la edad. Además, su fenotipo incluye malformaciones craneofaciales, ano-rectales, y de las extremidades y líneas de pigmentación anormales en la piel. Ocasionalmente se asocia con la presencia de hernia diafragmática y malformaciones cardíacas.5
Se reporta el caso de un niño diagnosticado con SPK por cariotipo convencional, reporte que es poco usual debido a las dificultades para realizar el diagnóstico de la anomalía cromosómica, ya que las pruebas citogenéticas convencionales usualmente pueden dar resultado normal, y solo en cultivo de tejidos puede demostrarse la presencia de la alteración cromosómica.
PRESENTACIÓN DEL CASO
Paciente hijo de madre de 34 años, producto de segunda gestación, de padres no consanguíneos, y antecedente materno de embarazo espontáneo. La madre realiza control prenatal mensual, con seguimiento ecográfico prenatal (3 ecografías), las cuales no reportaron malformaciones congénitas; niega exposición a drogas, alcohol o tabaco durante el embarazo.
Al momento del nacimiento, se reporta peso de 3 400 g y longitud 54 cm. Se evidencia la presencia de una malformación ano-rectal tipo ano imperforado bajo, macrocefalia sin hidrocefalia, polidactilia postaxial (Fig. 1) y reflujo vesiculoureteral.
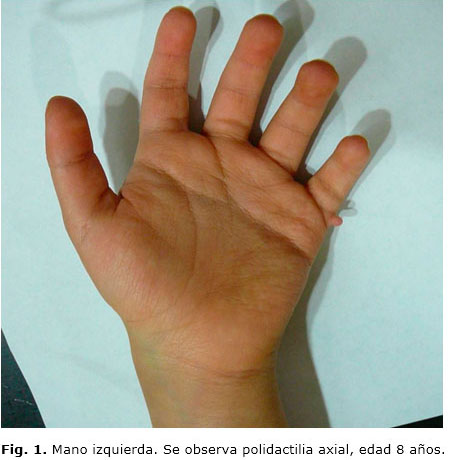
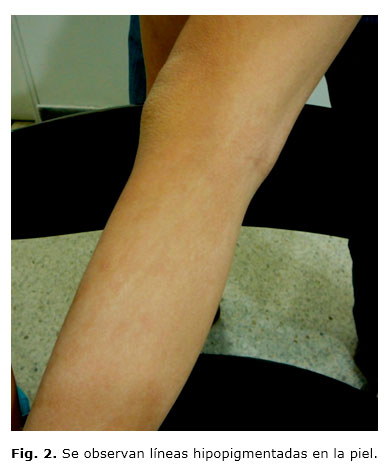
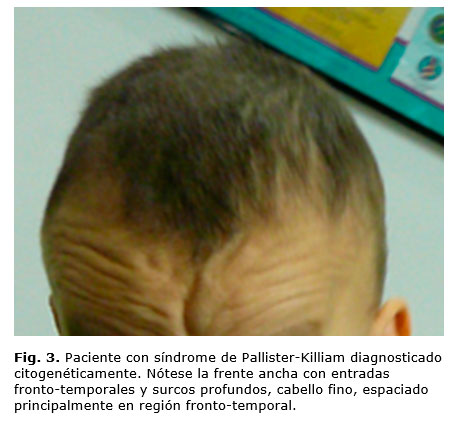
El paciente es remitido a la consulta de Genética Pediátrica a los 8 años de edad, y presenta, adicionalmente, retraso en el desarrollo psicomotor y retardo mental severo, dolicocefalia, movimientos repetitivos, hipotonía severa, manchas lineales hipopigmentadas (Fig. 2) y características faciales especiales, como mejillas llenas, labio inferior grueso, paladar ojival, alopecia fronto-temporal (Fig. 3); así como medidas antropométricas talla, peso, perímetro cefálico y medidas craneofaciales entre percentiles 75-95 para la edad. Estudios imagenológicos adicionales (resonancia magnética cerebral) evidenciaron agenesia de cuerpo calloso y megacisterna magna.
Se realiza cariotipo bandeo G en sangre periférica, en el cual se solicitó el análisis de 100 metafases por la sospecha de SPK, y el resultado evidenció la presencia de un isocromosoma 12p (Fig. 4).

DISCUSIÓN
El SPK es una anomalía cromosómica poco usual, cuyo diagnóstico se dificulta por su fenotipo variado, debido a que la anomalía no se encuentra en todas las células, y que el porcentaje de células afectadas tiende a disminuir con la edad.6 Usualmente, la anomalía se halla en el 0-2 % de los linfocitos, en el 50-100 % de fibroblastos y en el 100 % de amniocitos y células de la médula ósea en recién nacidos.7 La proporción de células tetrasómicas en linfocitos y fibroblastos no se relaciona con la gravedad de la expresión fenotípica, y es mayor en los fibroblastos de las áreas de la piel con alteraciones en la pigmentación.5,8 En el presente caso se logró identificar la alteración cromosómica presente en el SPK, lo cual permitió, no solo confirmar el diagnóstico clínico, sino descartar otras alteraciones cromosómicas detectadas por cariotipo convencional.
A pesar de que ninguna característica fenotípica es patognomónica de este desorden, su cuadro clínico se considera distintivo. Entre las características más destacadas se encuentran: presencia de facies tosca, anomalías en la pigmentación de la piel, alopecia fronto-temporal (hallazgo por el cual se sospechó el síndrome), retraso mental severo, y en algunas ocasiones, hernias diafragmáticas.5 Esta última condición es una de las causas más frecuentes de muerte posnatal temprana, lo que impide el diagnóstico de los neonatos con esta malformación, pues fallecen antes de poder realizarse un cariotipo.9,10
Los indicadores prenatales del SPK son: fémures cortos, polihidramnios y hernia diafragmática.11 Si estos indicadores están presentes, se recomienda hacer una amniocentesis, que puede ser muy útil para descartar un mosaicismo.12 La presencia de anomalías en la pigmentación sugiere la presencia de un mosaicismo, por lo cual, si este se encuentra, es pertinente la realización de un cariotipo en fibroblastos de las porciones de piel hipo o hiperpigmentadas.13,14
En cuanto al pronóstico neurológico se describe discapacidad intelectual con retardo motor severo, que se manifiesta a edades muy tempranas.5,12 Se ha asociado, en algunas ocasiones, a edad materna avanzada; sin embargo, el riesgo de recurrencia es muy bajo,2 por lo que debe tenerse en cuenta en el momento de la asesoría.
El diagnóstico diferencial de este síndrome incluye el síndrome de Fryns, por la presencia de hernia diafragmática; el síndrome orofaciodigital tipo IX, por algunas similitudes fenotípicas, pero tiene también hamartomas y anomalías oculares; el síndrome de Simpson Golabi Behmel, que presenta sobrecrecimiento, polidactilia, y facies tosca como hallazgo común con el síndrome de Beckwith Wiedemann, que tiene sobrecrecimiento, organomegalia, onfalocele y macroglosia; y el síndrome de Pallister Hall, también con retraso del neurodesarrollo, hamartomas y polidactilia. Esos diagnósticos pueden ser descartados con el estudio citogenético, por lo que el uso de esta herramienta diagnóstica resulta necesaria.
REFERENCIAS BIBLIOGRÁFICAS
1. Priest JH, Rust JM, Fernhoff PM. Tissue specificity and stability of mosaicism in Pallister-Killian + i(12p) syndrome: relevance for prenatal diagnosis. Am J Med Genet. 1992;42:820-4.
2. Wenger SL, Steele MW, Yu WD. Risk effect of maternal age in Pallisteri (12p) syndrome. Clin Genet. 1988;34:181-4.
3. Pallister PD, Meisner LF, Elejalde BR, Francke U, Herrmann J, Spranger J, et al. The Pallister mosaic syndrome. Birth Defects Orig Artic Ser. 1977;13:103-10.
4. Killian W, Teschler-Nicola M. Case report 72: mental retardation, unusual facial appearance, abnormal hair. Synd Ident. 1981;7:6-7.
5. Schinzel A. Tetrasomy 12p (Pallister-Killian syndrome). J Med Genet. 1991;28:122-5.
6. Peltomaki P, Knuutila S, Ritvanen A, Kaitila I, de la Chapelle A. Pallister-Killian syndrome: cytogenetic and molecular studies. Clin Genet. 1987;31:399-405.
7. Ward BE, Hayden MW, Robinson A. Isochromosome 12p mosaicism (Pallister-Killian syndrome): newborn diagnosis by direct bone marrow analysis. Am J Med Genet. 1988;31:835-9.
8. Bielanska MM, Khalifa MM, Duncan AMV. Pallister-Killian syndrome: a mild case diagnosed by fluorescence in situ hybridization. Review of the literature and expansion of the phenotype. Am J Med Genet. 1966;65:104-8.
9. Bergoffen JA, Punnett H, Campbell TJ, Ross AJ III, Ruchelli E, Zackai EH. Diaphragmatic hernia in tetrasomy 12p mosaicism. J Pediatr. 1993;122:603-6.
10. Jones KL. Smith’s recognizable patterns of human malformations. 6th. ed. Montreal: Elsevier; 2006. p. 263.
11. Wilson RD, Harrison K, Clarke LA, Yong SL. Tetrasomy 12p (Pallister-Killian syndrome): ultrasound indicators and confirmation by interphase FISH. Prenatal Diagn. 1994;14:787-92.
12. Day-Salvatore D, Smulian J, Guzman E, Mohan C, Weinberger B, Hanley ML. Genetics casebook. Pallister-Killian syndrome. J Perinatol. 1996;16:406-12.
13. Sybert VP. Hypomelanosis of Ito: a description, not a diagnosis. J Invest Dermatol. 1994;103(suppl 5):141S-3S.
14. Ritter CL, Steele MW, Wenger SL, Cohen BA. Chromosome mosaicism in hypomelanosis of Ito. Am J Med Genet. 1990;35:14-7.
Recibido: 11 de julio de 2014.
Aprobado: 4 de marzo de 2015.
Carolina Isaza de Lourido. Universidad del Valle. Dirección postal 760021. Cali, Colombia.
Correos electrónicos: hcarolinaisadel@gmail.com hmpachajoa@icesi.edu.co

