










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr vol.87 no.4 Ciudad de la Habana oct.-dic. 2015
Rev Cubana Pediatr. 2015;87(4)
ARTÍCULO ORIGINAL
Fibromatosis agresiva en la infancia en el servicio de Oncopediatría
Aggresive fibromatosis in childhood at the oncologic pediatric service
Dra. Migdalia Pérez Trejo, Dra. Mariuska Forteza Sáez, Dr. Jesús de los Santos Renó Céspedes, MSc. Débora García Socarrás, MSc. Dayne Quintero Vázquez, MSc. Idelmis Curbelo Heredia
Servicio de Oncopediatría. Instituto Nacional de Oncología y Radiobiología (INOR). La Habana, Cuba.
RESUMEN
Introducción: la fibromatosis abarca un amplio espectro de lesiones fibrosas proliferativas con apariencia microscópica similar, que afectan a diferentes localizaciones anatómicas. Se agrupan dentro de los tumores fibrosos benignos en niños, y poseen un potencial intermedio entre las lesiones benignas y malignas.
Objetivo: describir las características clínicas y el tratamiento de los pacientes con diagnóstico de fibromatosis agresiva tratados en el servicio de Oncopediatría en el Instituto Nacional de Oncología y Radiobiología.
Métodos: se realizó un estudio descriptivo, longitudinal y retrospectivo desde el 1º de enero de 2003 al 31 de diciembre de 2013, según variables demográficas, clínicas y terapéuticas. Se identificaron los pacientes a partir de las bases de datos del registro hospitalario del Instituto Nacional de Oncología y Radiobiología. Se seleccionaron todos los pacientes con diagnóstico histológico de esta enfermedad.
Resultados: se identificaron 9 pacientes con predominio del sexo masculino (56 %), con un rango de edades entre 0 y 9 años; y la localización más frecuente fue cabeza y cuello. Las modalidades de tratamiento utilizadas fueron: cirugía en 100 % de los casos, y quimioterapia y radioterapia concurrente (33 %). En estos momentos se cuenta con 100 % de supervivencia.
Conclusiones: la fibromatosis agresiva son lesiones benignas muy raras, agresivas localmente y sin potencial metastásico. Su tratamiento fundamental es la cirugía, sin embargo, deben incluirse otras modalidades terapéuticas para lograr el control local de la enfermedad.
Palabras clave: fibromatosis agresiva, tumor desmoide, cabeza y cuello.
ABSTRACT
Introduction: fibromatosis covers a wide spectrum of proliferative fiber lesions with similar microscopic appearance that affect various anatomical locations. These lesions are grouped into the benign fiber tumors in children and have an intermediate potential between the benign and the malignant lesions.
Objective: to describe the clinical characteristics of and the treatment prescribed for patients with diagnosis of aggressive fibromatosis, who were treated at the oncologic pediatrics service of the National Institute of Oncology and Radiobiology.
Methods: retrospective, longitudinal and descriptive study conducted from January 1st, 2003 through December 31st 2013 based on demographic, clinical and therapeutic variables. The patients were identified according to databases from the hospital register of the National Institute of Oncology and Radiobiology. All the patients with histological diagnosis for the disease participated in the study.
Results: nine patients were detected with predominance of males (56 %), age ranging from 0 to 9 years and the most common location were head and neck. The treatment modalities included surgery in 100 % of cases and concurrent chemotherapy and radiotherapy (33 %). Currently, the survival rate is 100 %.
Conclusions: aggressive fibromatosis are benign lesions that are very unusual, locally aggressive and with no metastatic potential. The main treatment is surgery; but other therapeutic variants should be included to achieve the local management of disease.
Keywords: aggressive fibromatosis, desmoids tumor, head and neck.
INTRODUCCIÓN
La fibromatosis agresiva —también denominada tumor desmoide— es un tumor benigno poco común, que se origina de las estructuras músculo-aponeuróticas profundas y/o del periostio; y son localmente agresivas, con tendencia a la recidiva local. No suele dar metástasis a distancia, pero pueden infiltrar estructuras vitales, y comprometer la vida del enfermo.1,2
Comprende el 0,03 % de las neoplasias en general y el 3 % de todos los tumores de partes blandas.3,4 Se estima una incidencia de 2 a 4 casos por cada millón de habitantes, y se diagnostican aproximadamente 900 nuevos casos anualmente en los Estados Unidos.5
El término fibromatosis fue propuesto originalmente por Stout en 1954 para referirse a un grupo de tumores que poseen fibroblastos proliferantes, miofibroblastos y marcado colágeno.6 Su etiología es desconocida, se ha asociado a cirugías y/o traumatismos previos, embarazo, tratamiento con estrógenos y síndrome de Gardner.7
La edad más frecuente de aparición es durante la primera década de la vida, pero hay casos descritos de todas las edades. Las localizaciones más frecuentes son: abdominal (57 %), extra abdominal (43 %), sobre todo en pelvis, tórax, mediastino, y cabeza y cuello. Clínicamente, se presentan como masas indoloras, aunque en ocasiones, dependiendo de la localización, pueden ser dolorosas.
Su diagnóstico es histológico y el tratamiento fundamentalmente quirúrgico.8 Se realizó el presente estudio con el objetivo de describir las características clínicas y el tratamiento terapéutico de los pacientes con diagnóstico de fibromatosis agresiva, en el Instituto Nacional de Oncología y Radiobiología (INOR), en el servicio de Oncopediatría, teniendo en cuenta la baja incidencia de esta enfermedad.
MÉTODOS
Se realizó un estudio descriptivo, longitudinal y retrospectivo. El universo consistió en todos los pacientes tratados con enfermedades malignas en el servicio de Oncopediatría del INOR; y la muestra estuvo conformada por los 9 pacientes con diagnóstico histológico de fibromatosis agresiva, desde el 1º de enero de 2003 al 31 de diciembre de 2013, con previa aprobación del consentimiento informado por parte de los padres o tutores legales en cada caso.
Las variables evaluadas fueron: edad, sexo, localización, presentación clínica, estudios de imagen (radiografía simple y tomografía axial computarizada [TAC] y/o resonancia magnética [RM]) de la zona afectada, así como diagnóstico por biopsia con estudio inmunohistoquímico en todos los casos y modalidad de tratamiento.
Para las variables estudiadas se utilizó el análisis de tablas de contingencia mediante el test exacto de Fisher, con un intervalo de confianza del 95 %. Los resultados se presentaron en tablas y figuras. Los datos fueron procesados en Excel y en el paquete estadístico SPSS versión 15.0.
RESULTADOS
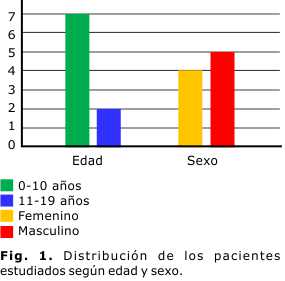
Se evaluaron un total de 510 pacientes pediátricos con enfermedades malignas en el período seleccionado; de ellos, 9 presentaron diagnóstico de fibromatosis agresiva, con ligero predominio del sexo masculino (56 %), con una edad media de 9 años (rango entre 0 y 9 años), y solo el 22 % eran mayores de 11 años (Fig. 1).
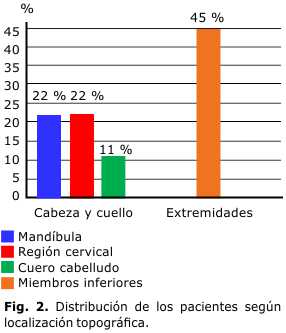
Las localizaciones fueron: cabeza y cuello (región cervical, mandíbula y cuero cabelludo), que representan el 55 %, seguido de zona glútea y miembros inferiores (Fig. 2).
No se presentaron síntomas graves, y la forma de presentación predominante fue la masa tumoral palpable de rápido crecimiento, seguido de la impotencia funcional (Fig. 3).

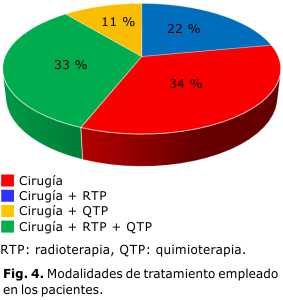
A todos los pacientes se les realizaron estudios de imágenes. El diagnóstico se realizó mediante biopsia incisional de la lesión en todos los pacientes. La cirugía exclusiva se empleó solo en 3 pacientes, y en el resto de los casos se combinó la cirugía con radioterapia (RTP) y quimioterapia (QTP) (Fig. 4). En estos momentos contamos con una supervivencia del 100 %.
DISCUSIÓN
En el período analizado se identificaron 9 pacientes con el diagnóstico de fibromatosis agresiva, confirmada histopatológicamente. La mayor incidencia fue antes de los 10 años de edad, con un rango de 2 a 17 años, resultado que coincide con los obtenidos por diferentes autores, en los cuales existe un pico de incidencia en la infancia a los 8 años (rango de edad 0-19 años), aunque puede aparecer en cualquier etapa de la vida.7 Se encontró predominio del sexo masculino, lo cual no coincide con los estudios revisados, que plantean un ligero predominio en mujeres.9,10
En la serie estudiada se encontró que el 55 % de los casos presentaban localizaciones extraabdominales, específicamente en el área de cabeza y cuello, mientras en el resto se afectaron zonas poco habituales, como extremidades inferiores. Este resultado coincide con las series revisadas, en las que la localización extraabdominal ocupa más de la mitad de los casos; por lo general, se asocia a casos de tipo esporádico, pueden presentarse en cualquier parte del cuerpo, aunque son más frecuentes en hombros, pared torácica y parte superior de los brazos.
Se describe que el 15 % de todos los tumores desmoides extraabdominales ocurren en el área de cabeza y cuello, y sus localizaciones más comunes son: cuello, laringe, senos paranasales, fosa infratemporal y cavidad oral. Puede presentarse como una enfermedad multicéntrica (10 % de los casos), y la aparición de las lesiones puede ser sincrónica o meta-crónica.1,10 Masson y Soule recogen 284 casos en la “Clínica Mayo”, y 83 casos registrados por el “Johns Hopkins”,11 y el orden de frecuencia es: abdomen, pelvis, tórax, cabeza, cuello y mediastino. El 10 % de las fibromatosis extraabdominales afectan a la región anatómica de cabeza y cuello, localización más común en la edad pediátrica, según la American Academy of Oral Pathology,12 y que coincide con nuestros resultados.
La manifestación clínica más frecuente fue la masa tumoral palpable en el área de cabeza y cuello, de crecimiento progresivo, no doloroso y fijo a planos profundos. Por su localización puede asociarse a síntomas como trismo, disnea, disfagia o proptosis, que dependerá del área que se encuentre afectando.1 Radiológicamente posee una apariencia infiltrativa con respecto al músculo esquelético. La TAC y la RM pueden mostrar un tumor isodenso o hiperintenso con respecto al músculo esquelético.10
El diagnóstico histológico se realizó en todos los casos mediante biopsia incisional, independientemente de su localización. Estas lesiones proliferativas son histológicamente similares: pequeños paquetes de células fusiformes, con un estroma fibroso abundante, celularidad variable pero baja, fascículos de fibroblastos hipocelulares y miofibroblastos careciendo de núcleos, que muestran una pequeña actividad mitótica con necrosis ausente.1,5 Cuando se les realiza estudios de inmunohistoquímica suelen marcar vimentina, c-kit y receptores de estrógenos principalmente; sin embargo, puede marcar receptores de progesterona, her2-neu, ki-67, entre otros.13
Debe hacerse diagnóstico diferencial con fibrosarcoma, neurofibroma, fascitis nodular, histiocitoma fibroso y miofibromatosis infantil. Ocasionalmente los tumores desmoides suelen sobrediagnosticarse como fibrosarcomas (los cuales poseen un patrón de crecimiento desordenado, mayor cantidad de células inmaduras y de mitosis, además de la posibilidad de metástasis a distancia), o subdiagnosticarse como fibrosis reactiva, la cual se presenta generalmente después de un trauma o lesión.9 Una vez que se tenga claro que se trata de una fibromatosis extraabdominal, comienza el dilema para su tratamiento. Particularmente en el área de cabeza y cuello, la dificultad en la toma de decisiones es debido a la cercanía a estructuras anatómicas vitales, por lo que se sugiere que la terapéutica siempre debe ser multimodal; sin embargo, debido a su poca frecuencia no hay lineamientos establecidos para su tratamiento.1
La cirugía sola con margen oncológico se realizó en 3 pacientes, y en 2 de ellos fue necesario realizar la amputación del miembro inferior, técnica que debe ser reservada para aquellos pacientes con múltiples recurrencias, en los cuales no es factible realizar cirugía conservadora, por lo extenso de la lesión o la invasión de estructuras vasculares o nerviosas vecinas. A pesar de lo controversial del tratamiento, hasta el momento actual todos los autores coinciden, en que este es el tratamiento inicial de elección.1,2 En el resto de los pacientes, debido a la localización de la lesión, fue necesario el uso de modalidades combinadas de tratamiento, como la RTP posoperatoria para el control de la enfermedad microscópica residual.
La dosis habitual es de aproximadamente 50 Gy.1,2 La RTP puede darse también como tratamiento único. Hay algunos estudios que demuestran que ella sola puede lograr respuestas completas y parciales de hasta de 40 %, y conseguir mantener una enfermedad estable en 53 %, lo cual pudiera evitar la necesidad de realizar grandes cirugías.14 Sin embargo, el criterio de usar la RTP sola se aplica, principalmente, en los casos que el tumor sea considerado irresecable.2 En nuestro trabajo no se empleó como tratamiento único en ninguno de los casos.
Una pauta de tratamiento adecuada es la propuesta por la Sociedad Internacional de Oncología Pediátrica (SIOP), la cual considera, que la RTP en niños estaría contraindicada, por el riesgo de inducir la malignización del tumor, y porque las altas dosis requeridas (45-60 Gy) para conseguir el control de la enfermedad, acarrearían graves secuelas.15
En uno de los pacientes afectados se utilizó terapia hormonal con tamoxifén, cuya efectividad se describe en hasta el 50 %. Si el tumor expresa receptores estrogénicos supone una mayor efectividad, aunque se describe cierta respuesta aunque no exprese este receptor.13,15
La terapia sistémica ha probado ser efectiva en los pacientes con múltiples recurrencias locales, al igual que en pacientes con tumores considerados irresecables, o tumores intraabdominales.16 Uno de los tratamientos más usados son los antiinflamatorios no esteroideos (AINES); y entre los más efectivos se encuentran los inhibidores de la enzima COX-1, como la indometacina y el sulindac, y los inhibidores de la COX-2, como el meloxicam.15,17
En 3 pacientes se utilizó la QTP citotóxica como complemento a la cirugía, que puede desempeñar un papel importante en el tratamiento de los tumores no resecables, o con resecciones consideradas marginales, y en aquellos pacientes con progresión o recurrencia de la enfermedad. Se añade como alternativa a la cirugía —si la resección total no es posible y el tumor progresa— el uso de QTP con vincristina y actinomicina-D. También se han publicado revisiones con pautas de QTP tipo VAC: vincristina (VCR), ciclofosfamida (CFM) y actinomicina (ACTD), ocasionalmente ampliada con adriamicina, con buenos resultados.18,19 Posterior al tratamiento con cirugía sola, e inclusive si los márgenes de resección han sido negativos, se espera una recurrencia local de hasta el 20 %, mientras que si se reportan márgenes positivos, se eleva al 68 %. Ahora bien, si el paciente recibe RTP posoperatoria, la posibilidad de recaída disminuye significativamente. El 80 % de las recurrencias aparecen generalmente los 2 primeros años después del tratamiento.2,3
En conclusión, la edad más frecuente de aparición de la fibromatosis agresiva es en menores de 10 años, con ligero predominio en el sexo masculino. La masa tumoral palpable resulta la principal manifestación clínica, con una mayor localización en cabeza y cuello. El tratamiento fundamental es la cirugía, sin embargo, deben incluirse otras modalidades terapéuticas para lograr el control local de la enfermedad. El número pequeño de la muestra no permite hacer inferencias estadísticas.
REFERENCIAS BIBLIOGRÁFICAS
1. Sobani ZA, Junaid M, Khan MJ. Successful management of aggressive fibromatosis of the neck using wide surgical excision: A case report. J Med Case Rep. 2011;5:244.
2. Francisco JL, Garriga E, Dacunha M, Tirado E, Siso S, Brito E. Fibromatosis agresiva extra abdominal de cabeza y cuello. Rev Venez Oncol. 2014;26(3):217-22.
3. Baumert BG, Spahr MO, Von Hochstetter A, Beauvois S, Landmann C, Fridrich K, et al. The impact of radiotherapy in the treatment of desmoid tumours: An international survey of 110 patients. A study of the rare cancer network. Radiat Oncol. 2007;2:12.
4. Lakhan SE, Eager RM, Harle L. Aggressive juvenile fibromatosis of the paranasal sinuses: Case report and brief review. J Hematol Oncol. 2008;1:3.
5. Liu Y, Guan GF, Jin CS, Yang JP. Aggressive fibromatosis of the larynx: Case report and Brief Review. J Int Med Res. 2011;39(2):682-9.
6. Kohli K, Kawatra V, Khurana N, Jain S. Multicentric synchronous recurrent aggressive fibromatosis. J Cytol. 2012;29(1):57-9.
7. Contreras MA, Ruiz PR, Martínez MN, Galeas FJ, Ruiz FD, Valiente AÁL, et al. Fibromatosis agresiva de cabeza y cuello en la edad pediátrica. Un caso clínico y revisión de la literatura. Cir Pediatr. 2012;25:213-7.
8. Dufresne A, Bertucci F, Penel N, Le Cesne A, Bui B, Tubiana-Hulin M, et al. Identification of biological factors predictive of response to imatinib mesylate in aggressive fibromatosis. Br J Cancer. 2010;103(4):482-5.
9. Shetty DC, Urs AB, Ahuja P, Sikka S. Aggressive fibromatosis of anterior maxilla. J Oral Maxillofac Pathol. 2011;15(1):85-7.
10. Vida LP, Martínez FR. Tumores desmoides intraabdominales. Med Clin (Barc). 2013;141(7):314-9.
11. Valero HF, Montaner DA, Peralta NJ. Fibromatosis palmar infantil: a propósito de un caso. Revista Española de Cirugía Osteoarticular. 2014;49(259):155-8.
12. Docampo J, Santoro D, Bruno C, Morales C. Fibromatosis orbitaria solitaria. Reporte de caso. Revista Argentina Radiología. 2010;74(1):43-6.
13. Leithner A, Gapp M, Radl R, Pascher A, Krippl P, Leithner K, et al. Immunohistochemical analysis of desmoids tumors. J Clin Pathol. 2005;58(11):1152-6.
14. Rudiger HA, Ngan SY, Ng M, Powell GJ, Choong PF. Radiation therapy in the treatment of desmoid tumors reduces surgical indications. Eur J Surg Oncol. 2010;36(1):84-8.
15. Hawkins DS, Spunt SL, Stephen XS. Children’s Oncology Group’s 2013 Blueprint for Research: Soft Tissue Sarcomas. Pediatr Blood Cancer. 2013 June;60(6):1001-8.
16. Ruiz-Osuna C, Ávila-Zamorano ML, López-Durán A. Fibromatosis agresiva infantil de cadera con destrucción articular grave. Acta Ortopédica Mexicana. Jul-Ago 2010;24(4):267-72.
17. Nishida Y, Tsukushi S, Shido Y, Wasa J, Ishiguro N, Yamada Y. Successful treatment with meloxicam, a cyclooxygenase-2 inhibitor, of patients with extraabdominal desmoid tumors: A pilot study. J Clin Oncol. 2010;28(6):e107-9.
18. Chugh R, Chugh R, Wathen JK, Patel SR, Maki RG, Meyers PA, et al. Sarcoma Alliance for Research through Collaboration (SARC): Efficacy of imatinib in aggressive fibromatosis: Results of a phase II multicenter Sarcoma Alliance for Research through Collaboration (SARC) trial. Clin Cancer Res. 2010;16(19):4884-9481.
19. Fernández GR, Sangüesa MJ, Villanueva GE. Fibromatosis extra-abdominal agresiva. Descripción de un caso y revisión de la literatura. Revista Española de Cirugía Osteoarticular. 2009;45(238):86-92.
Recibido: 14 de mayo de 2015.
Aprobado: 13 de junio de 2015.
Migdalia Pérez Trejo. Instituto Nacional de Oncología y Radiobiología (INOR). Calle 29, esquina F, Vedado, municipio Plaza de la Revolución. La Habana, Cuba.
Correo electrónico: mforteza@infomed.sld.cu

