










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr vol.89 no.2 Ciudad de la Habana abr.-jun. 2017
Rev Cubana Pediatr. 2017;89(2)
ARTÍCULO ORIGINAL
Caracterización clínica y genética de la fibrosis quística en la provincia de Holguín
Clinical and genetic characterization of cystic fibrosis in Holguin province
Elayne Esther Santana Hernández,I Víctor Jesús Tamayo Chang,I Teresa Collazo Mesa,II Ixchel López Reyes,II Fabio Feria Estrada,III Fidel Rodríguez CalaIV
ICentro Provincial de Genética Médica. Holguín, Cuba.
IICentro Nacional de Genética Médica. La Habana, Cuba.
IIIHospital Pediátrico Provincial "Octavio de la Concepción de la Pedraja". Holguín, Cuba.
IVHospital Pediátrico "William Soler". La Habana, Cuba.
RESUMEN
Introducción: la fibrosis quística es una enfermedad genética caracterizada por alteraciones pulmonares crónicas, insuficiencia pancreática exocrina y una elevada concentración de los electrólitos en el sudor. Es causada por la presencia de mutaciones en el gen CFTR, que codifica para un canal de cloro denominado proteína reguladora de la conductancia transmembranal (CFTR), localizado en el cromosoma 7. Se hereda con carácter autosómico recesivo, y expresa gran heterogeneidad clínica y genética.
Objetivo: describir las principales características clínicas y genéticas que presentan los enfermos con fibrosis quística en la provincia de Holguín.
Método: se realizó un estudio descriptivo, tipo serie de casos, en el que la muestra estuvo formada por 22 pacientes con diagnóstico clínico y molecular de fibrosis quística en la provincia de Holguín, en el período de Enero de 2009 a Enero de 2016.
Resultados: predominó el sexo masculino, con 15 afectados entre 6 a 10 años, el 81,8 % de los pacientes presentó manifestaciones respiratorias crónicas, y el signo clínico más frecuentemente asociado fue la desnutrición en 14 enfermos (63,6 %). Los heterocigóticos compuestos totalizaron el 59,09 %, los homocigóticos el 40,9 %, la mutación ΔF508 en forma homocigótica se presentó en solo 4 afectados, y heterocigótico compuesto en 7, para 31,8 %.
Conclusiones: la mutación más frecuentemente encontrada es la deleción ΔF508, y la enfermedad respiratoria crónica la manifestación clínica que predominó. Los pacientes homocigóticos a la mutación ΔF508 están más afectados desde el punto de vista clínico.
Palabras clave: fibrosis quística; mucoviscidosis; glándulas exocrinas; gen CFTR; proteína CFTR; mutaciones; polimorfismos.
ABSTRACT
Introduction: cystic fibrosis is one type of genetic disease characterized by chronic pulmonary alterations, exocrine pancreatic failure and high concentration of electrolytes in sweat. It is caused by mutations in CFTR gene that codes for a chlorine channel called transmembrane conductance regulator protein (CFTR) located in chromosome 7. It is a hereditary recessive autosomal disease and expresses great clinical and genetic heterogeneity.
Objective: to describe the main clinical and genetic characteristics of patients with cystic fibrosis in Holguin province.
Methods: a case-series descriptive study was conducted in a sample of 22 patients with clinical and molecular diagnosis of cystic fibrosis from Holguin province in the period of January 2009 through January 2016.
Results: males predominated, 15 diagnosed patients aged 6 to 10 years, 81.8 % of the sample presented with chronic respiratory manifestations and the most frequently associated sign was malnutrition in 14 patients (63.6 %). The compound heterozygotes accounted for 59.09 % and the homozygotes for 40.9 %; homozygotic ΔF508 mutation affected 4 patients whereas compound heterozygotic form was seen in 7, representing 31.8 %.
Conclusions: the most found mutation is ΔF508 deletion and the predominant clinical manifestation is chronic respiratory disease. The patients with homozygotic ΔF508 mutation were the most clinically affected persons.
Keywords: cystic fibrosis; mucoviscidosis; exocrine glands; CFTR gene; CFTR protein; mutations; polymorphisms.
INTRODUCCIÓN
En 1936 el pediatra suizo Guido Fanconi fue el primero en usar el término fibrosis quística (FQ) para describir la combinación de insuficiencia pancreática y enfermedad pulmonar crónica en niños, citado por Rojo y Collazo.1,2 En 1938 Dorothy Andersen asoció íleo meconial con FQ, y describió este trastorno separado de la enfermedad celíaca, citado por Collazo.3 En el mismo año, Blackfan y otros, describen 35 niños con atrofia y fibrosis del páncreas debido a espesamiento de secreciones.4 En 1943, se reconoce a la FQ como una enfermedad sistémica, y se acuña el término "mucoviscidosis", citado por Barrado y Cutting.5,6
En 1945, un primer estudio declara 46 familias afectadas, y otro estudio informó 56 familias más, ambos concluyen que la FQ se hereda en forma autosómica recesiva, citado por Lucarelli.7 En 1953, Paul Di Sant'Agnese asigna valor diagnóstico a los electrolitos del sudor; y en 1989, el grupo de Lap-Chee Tsui identifica y clona el gen de la FQ, ubicado en el brazo largo del cromosoma 7, y a la proteína para la cual codifica, la denomina proteína de conductancia transmembranal de la FQ (CFTR, por sus siglas en inglés), publicando los hallazgos en un memorable número de la revista Science, citado por Lucarrelli,7 Cholon8 y Sosnay.9
La FQ, o mucoviscidosis, es el padecimiento autosómico recesivo más frecuente en la población caucásica, en la cual se presenta con una incidencia de 1 en cada 2 000-3 500 recién nacidos vivos, y se estima que 1 de cada 25 individuos es portador sano de la mutación, citado por Knowles10 y Rowe.11
En países desarrollados los pacientes con FQ tienen una expectativa de vida de aproximadamente 37 años, y de 50 años, como exhibe Dinamarca. En Suecia la frecuencia es de 1 por 2 200 y 1 por 4 100 recién nacidos vivos; en Canadá, entre 1 por 891 y 1 por 2 700 recién nacidos vivos. En América Latina la incidencia varía en los diferentes países: 45 % en México, 50,8 % en Brasil, entre 57 y 60,9 % en Argentina, en Uruguay es de 40 %, y en Cuba se reporta el 34 %, citado por Melotti,12 Sands13 y Alonso.14 Es considerada la más letal de las afecciones crónicas hereditarias del pulmón. Se plantea que en el mundo existen alrededor de 66 mil enfermos. Su frecuencia varía desde 1 x 2 500 nacimientos en poblaciones blancas, hasta 1 × 15 000 en la raza negra y 1 × 31 000 en asiáticos, citado por Fernández,15 Brodlie16 y Razon.17
El primer caso se reporta en Cuba en 1953, en una autopsia, por el anatomopatólogo cubano Salas Panisello, de la Universidad de La Habana, citado por Rojo y Collazo.1,2 La doctora Borbolla, en 1962, describe un paciente, y el doctor Mir del Junco otro en 1965, citado por Collazo.3 En 1970, el doctor Manuel Rojo Concepción y sus colaboradores, reportan 3 casos en la Revista Cubana de Pediatría; y más tarde, en el Congreso Internacional de FQ de 1974, citado por Rojo,1 el profesor presenta la casuística de un grupo de pacientes estudiados en el hospital "Pedro Borrás", citado por Rojo y Collazo.1-4
En 1974 se crea la Comisión Nacional de FQ y se establece el Registro Cubano de FQ, incorporado al Registro Latinoamericano (REGLAF). En la década del 90 se reportan alrededor de 860 pacientes, de los cuales 122 son cubanos. Los registros documentan alrededor de 257 pacientes con FQ, de los cuales el 60 % corresponde al sexo masculino, y más del 50 % de estos sobrepasan los 15 años de edad.1,18-20
La frecuencia de la FQ en Cuba es algo menor de 1 x 5 000 nacidos vivos, y aproximadamente 1 de cada 25 personas es portadora de mutaciones para esta enfermedad. En el año 2015 existían en el país 273 pacientes con esta enfermedad, de los cuales el 61 % pertenecían al sexo masculino y el 39 % al sexo femenino, y con más de 19 años el 38,9 %, con un promedio de 13 diagnósticos por año, con una incidencia de 1:3 900, destacándose las provincias de La Habana, Santiago de Cuba y Holguín, citado por Fernández.15
La incidencia de esta enfermedad en la provincia nos motivó a realizar este estudio. Constituye una problemática de salud, y son necesarios estudios que caractericen a estos afectados y sus familiares, desde el punto de vista clínico y genético, para conocer los posibles matrimonios heterocigotos en riesgo de tener hijos enfermos. También poder brindar estudios prenatales a estas parejas, y realizar un adecuado asesoramiento genético a estas familias.
MÉTODOS
Se realizó un estudio descriptivo, tipo serie de casos. El universo estuvo formado por los 22 enfermos, coincidiendo con la muestra en estudio. Estos 22 pacientes con diagnóstico clínico y molecular de FQ, residentes en la provincia Holguín, se atendieron por consulta en el Hospital Pediátrico Provincial de Holguín, en el servicio de Genética Clínica, en el período de Enero de 2009 a Enero de 2016.
Se revisaron las historias clínicas para recoger datos de interés, como fue: sexo y la edad de los enfermos, y se agruparon en menores de un año, y los demás, cada 4 años. También se analizaron las manifestaciones clínicas, dentro de estas, el retardo del crecimiento, la desnutrición, la enfermedad respiratoria crónica, así como las manifestaciones digestivas y la insuficiencia pancreática.
Se les solicitó a los familiares el consentimiento informado (accedieron con firma por escrito su aceptación) para poder realizar extracción de sangre periférica para el examen de ADN, con el objetivo de realizar estudio molecular directo de las mutaciones, que se analizaron en el Centro Nacional de Genética Médica de La Habana, previo estudio de electrolitos en sudor positivo en 3 ocasiones. Se respetaron los principios éticos para estudios y así poder publicar estos resultados sin ofrecer su identidad.
El criterio de inclusión se estableció cuando se tratara de pacientes atendidos en consulta con manifestaciones sugestivas de FQ, que tuviesen electrolitos de sudor positivo y estudio molecular confirmado por mutaciones reconocibles estandarizadas en el Centro Nacional de Genética Médica. Se utilizó estadígrafos para medianas y por cientos para agrupar estos datos y saber el por ciento de cada uno de ellos.
RESULTADOS
Después de revisadas las historias clínicas, recogidos los datos clínicos relevantes y obtenidos los resultados de las mutaciones, se distribuyeron los 22 pacientes según su edad y sexo, hubo solo 3 menores de 1 año con la enfermedad, como se aprecia en la tabla 1. Se observa mayor número de enfermos en las edades comprendidas entre 6 a 10 años (27,2 %).
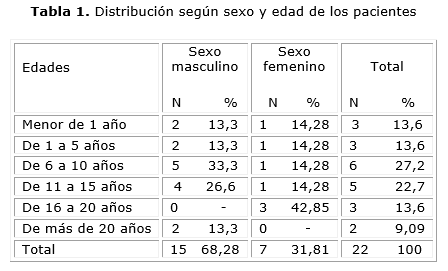
Dentro de las manifestaciones clínicas predominaron las manifestaciones respiratorias de forma crónica en 18 enfermos (81,8 %), como se representa en la tabla 2, seguidas por la desnutrición, en 14 afectados (signo clínico conocido y estudiado en estos pacientes). Sin embargo, la insuficiencia pancreática, se observó asociada al retardo del crecimiento y desarrollo en 4 pacientes (18,1 %), mientras que las manifestaciones digestivas se manifestaron en 2 enfermos.

En la tabla 3 se muestran las mutaciones encontradas en cada uno de estos pacientes. Como se observa, existen 9 pacientes con mutaciones de forma homocigótica y de estos, 4 con la mutación ΔF508. Estos están gravemente afectados, con un deterioro mayor de su función respiratoria, con insuficiencia crónica y un marcado retardo del crecimiento y desarrollo. Estos mismos son los que exhiben insuficiencia pancreática y desnutrición. Dentro de estos hay 11 que presentan mutaciones de forma heterocigótica, y de ellos 7 en combinación con la mutación ΔF508, que constituye la mutación por deleción de la proteína de transporte que está afectada en este gen CFTR en la posición 508.

DISCUSIÓN
La FQ es una enfermedad hereditaria con patrón de herencia autosómica recesiva, que expresa gran heterogeneidad clínica y genética, por su amplia heterogeneidad alélicas.1-3 La expresión heterogénea de la proteína CFTR evidencia que las diferentes mutaciones se manifiestan con una evolución clínica altamente variable.3
La combinación de los distintos tipos de mutación en un individuo y la evaluación de las características clínicas que presentan los pacientes, permite establecer las correlaciones entre el genotipo y el fenotipo. Incluso, antes de que se identificase el gen CFTR, se sabía que parte de las diferencias clínicas de los pacientes FQ están determinadas genéticamente. Alrededor del 15 % de los pacientes FQ son pancreáticos suficientes (PS), comparados con la mayoría de los pacientes con FQ, los cuales precisan del tratamiento con enzimas pancreáticas, pancreático insuficientes (PI), y quienes, por lo general, son los que más manifestaciones digestivas presentan.4-6
Cuando se revisa la bibliografía internacional se reconoce que la mutación ΔF508, representa el 66 % de los enfermos por FQ en todo el mundo. Esta mutación provoca una alteración en el proceso postranscripcional de la proteína, con una disminución del tráfico de la proteína madura, desde el aparato de Golgi a la superficie celular apical.7-9
Se conocen en la actualidad cerca de 2 000 mutaciones diferentes que ocasionan esta enfermedad, que se clasifican en 6 clases, en dependencia del defecto subyacente:10-13
- Clase I: déficit de síntesis.
- Clase II: defecto de maduración al pasar al retículo endoplásmico.
- Clase III: bloqueo de activación.
- Clase IV: defecto de la conducción.
- Clase V: empalme incorrecto.
- Clase VI: defecto de regulación.
Las mutaciones de clase I a III se asocian a fallas severas de producción del CFTR, y por lo tanto, a fenotipos más graves, en los que a la enfermedad respiratoria se asocia la insuficiencia pancreática con esteatorrea y malabsorción. La deleción de la fenilalanina en la posición 508 (ΔF508) es la mutación más frecuente (clase II).4,6 En este estudio, en los enfermos que presentan esta mutación, su función respiratoria está severamente afectada, como lo descrito en otras investigaciones.14-17
En poblaciones como la caucásica, en la cual la mutación ΔF508 es sumamente frecuente, en el gen CFTR (ΔF508, G542X) se detecta del 85-90 % de los alelos FQ. Estudios extensos para la identificación de marcadores moleculares, dentro y fuera del gen CFTR, han revelado que ciertas mutaciones como la ΔF508, G542X y N1303K se encuentran asociadas con haplotipos únicos, lo que sugiere que cada una de ellas se derivó de un mismo evento mutacional.18 Con base en el análisis de haplotipos se ha sugerido que la mutación ΔF508 se originó en Europa, mientras que la G542X fue introducida a España por los fenicios.19,20 Esto confirma que, debido a la colonización por lo europeos y los españoles de las Américas, estas dos mutaciones sean las de mayor frecuencia, como lo encontrado en este estudio.21,22
La FQ es una enfermedad genética frecuente en la raza caucásica, todos estos enfermos son de tez blanca y con antecedentes en sus familias de ancestros de inmigrantes españoles. En estas regiones la población era pequeña, y se mantuvo cerrada con una práctica social por mucho años, como fue la endogamia, que provocó muchos matrimonios consanguíneos, lo que pudiera explicar que se segregaran estas mutaciones de generación en generación, y aumentaran la tasa de heterocigóticos sanos, lo que, unido a la poca mezcla con otras regiones, explica que nazcan con mayor probabilidad enfermos en esta región del país. Esto sustenta más la hipótesis de que la frecuencia de portadores heterocigóticos sanos en la provincia es alta.
Estos pacientes son seguidos en consulta multidisciplinaria cada tres meses, en conjunto Pediatría, Genética, Fisiatría y Rehabilitación, Nutrición, Microbiología y Neumología. Se realiza valoración antropométrica de su función respiratoria y estudios microbiológicos, para evaluar sus ingresos, previo exudado faringeo, siguiendo el protocolo de antibióticos establecido nacionalmente, según el estadio clínico. Se encuentran 2 de estos pacientes en insuficiancia respiratoria crónica, con estadias prolongadas, e ingresos continuos sin mejoras sustanciales.
Se están realizando los estudios moleculares a los familiares de primer grado de estos pacientes, para conocer la cantidad de portadores sanos en cada una de estas familias, con el fin de brindar estudios prenatales moleculares en las parejas de riesgo. Se considera importante continuar estos estudios, para realizar un adecuado asesoramiento genético.
Se concluye que la mutación más frecuentemente encontrada es la deleción ΔF508, y la enfermedad respiratoria crónica la manifestación clínica que predominó. Los pacientes homocigóticos a la mutación ΔF508 están más afectados desde el punto de vista clínico.
Conflicto de intereses
Los autores declaran no tener conflicto de intereses en la realización del estudio.
REFERENCIAS BIBLIOGRÁFICAS
1. Rojo Concepción M, Quintero Enamorados I, Delgado Lopez H, Razón Behar R, Mir del Junco J, García Quesada M, et al. Cuban Commission of cystic fibrosis. Registration of patients since 1977 . Bol Med Hosp Infant Mex [serie en Internet]. 1980 Jul-Aug [citado 28 de Marzo de 2016];37(4). Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/7407012 .
2. Collazo T, Magarino C, Chavez R, Suardiaz B, Gispert S, Gomez M, et al. Frequency of delta-F508 mutation and XV2C/KM19 haplotypes in Cuban cystic fibrosis families. Hum Hered [serie en Internet]. 1995 Jan-Feb [citado 28 de Marzo de 2016];45(1). Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/7896301
3. Collazo T, Bofill AM, Clark Y, Hernández Y, Gómez M, Rodríguez F, et al. Common mutations in Cuban cystic fibrosis patients. J Cyst Fibros [serie en Internet]. 2009 Jan [citado 28 de Marzo de 2016];8(1). Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/18938114 .
4. Collazo T, López I, Clark Y, Piloto Y, González L, Gómez M, et al. Antenatal testing for cystic fibrosis in Cuba, 1988-2011. MEDICC Rev [serie en Internet]. 2014 Jul-Oct [citado 28 de Marzo de 2016];16(3-4). Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/25208115
5. Barrado L, Brañas P, Orellana MA, Martínez MT, García G. Molecular Characterization of Achromobacter Isolates from Cystic Fibrosis and Non-Cystic Fibrosis Patients in Madrid, Spain. J Clin Microbiol [serie en Internet]. 2013 June [citado 28 de Marzo de 2016];51(6). Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3716108/
6. Cutting GR. Cystic fibrosis genetics: from molecular understanding to clinical application. Nat Rev Genet [serie en Internet]. 2015 January [citado 28 de Marzo de 2016]. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4364438/
7. Lucarelli M,Bruno SM, Pierandrei S, Ferraguti G, Stamato A, Narzi F, et al. A Genotypic-Oriented View of CFTR Genetics Highlights Specific Mutational Patterns Underlying Clinical Macrocategories of Cystic Fibrosis. Mol Med [serie en Internet]. 2015 [citado 28 de Marzo de 2016];21(1). Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4503653/
8. Cholon DM, Quinney NL, Fulcher ML, Esther CR, Das Jr. J. Potentiator Ivacaftor Abrogates Pharmacological Correction of ΔF508 CFTR in Cystic Fibrosis. Sci Transl Med [serie en Internet]. 2014 July 23 [citado 28 de Marzo de 2016];6(246). Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4272825/
9. Sosnay PR, Siklosi KR, Goor FV, Kaniecki K, Yu H. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat Genet [serie en Internet]. 2013 October [citado 28 de Marzo de 2016];45(10). Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3874936/
10. Knowles MR, Drumm M. The Influence of Genetics on Cystic Fibrosis Phenotypes. Cold Spring Harb Perspect Med [serie en Internet]. 2012 December [citado 28 de Marzo de 2016];2(12). Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3543075/
11. Rowe SM, Heltshe SL, Gonska T, Donaldson SH, Borowitz D. Clinical Mechanism of the Cystic Fibrosis Transmembrane Conductance Regulator Potentiator Ivacaftor in G551D-mediated Cystic Fibrosis. Am J Respir Crit Care Med [serie en Internet]. 2014 July 15 [citado 28 de Marzo de 2016];190(2). Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4226057/
12. Melotti P, Mafficini A, Lebecque P, Ortombina M, Leal T, Pintani E, et al. Impact of MIF Gene Promoter Polymorphism on F508del Cystic Fibrosis Patients. PLoS One [serie en Internet]. 2014 [citado 28 de Marzo de 2016];9(12). Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4264759/
13. Sands D, Umławska W, Zielińska A. A cross-sectional study of growth, nutritional status and body proportions in children and adolescents at a medical center specializing in the treatment of cystic fibrosis in Poland. Arch Med Sci [serie en Internet]. 2015 March 16 [citado 28 de Marzo de 2016];11(1). Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4379371/
14. Alonso MJ, Heine-Sunyer D, Calvo M, Rosell J, Jiménez J, Ramos MD, et al. Spectrum of mutations in the CFTR qene in cystic fibrosis patients of Spanish ascestry. Ann Hum Genet [serie en Internet]. 2007 [citado 28 de Marzo de 2016];71. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed .
15. Fernández FM, Lara AF, Rodríguez Cala F, García Castañeda H, Fernández García S, Roblejo Balbuena H. Fibrosis quística. Diagnóstico tardío en el adulto presentación de caso. Rev Hab de Ciencias Médicas. 2010;9(2):196-202.
16. Brodlie M, Haq IJ, Roberts K, Elborn JS. Targeted therapies to improve CFTR function in cystic fibrosis. Genome Med [serie en Internet]. 2015 [citado 28 de Marzo de 2016];7(101). Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4582929/
17. Razón Behar R, Rodríguez Calá R, Rojo Concepción M, González Valdés JA, Abreu Suárez G, Pérez Rodríguez T, et al. La fibrosis quística en Cuba [homepage en Internet]. 2008 [citado 18 de Mayo de 2016]. Disponible en: http://www.sld.cu/galerias/pdf/sitios/pediatria/la_fibrosis_quistica_en_cuba.pdf
18. Gallati S. Disease-modifying genes and monogenic disorders: experience in cystic fibrosis. Appl Clin Genet [serie en Internet]. 2014 [citado 28 de Marzo de 2016];7. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4104546/
19. Ziętkiewicz E, Rutkiewicz E, Pogorzelski A, Klimek B, Voelkel K. CFTR Mutations Spectrum and the Efficiency of Molecular Diagnostics in Polish Cystic Fibrosis Patients. PLoS One [serie en Internet]. 2014 [citado 28 de Marzo de 2016];9(2). Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3935850/
20. Wang Y, Liu J, Loizidou A, Bugeja LA, Warner R. CFTR potentiators partially restore channel function to A561E-CFTR, a cystic fibrosis mutant with a similar mechanism of dysfunction as F508del-CFTR. Br J Pharmacol [serie en Internet]. 2014 October [citado 28 de Marzo de 2016];171(19). Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4209154/
21. Dong Q, Ernst SE, Ostedgaard SL, Shah VS, Ver Heul AR. Mutating the Conserved Q-loop Glutamine 1291 Selectively Disrupts Adenylate Kinase-dependent Channel Gating of the ATP-binding Cassette (ABC) Adenylate Kinase Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) and Reduces Channel Function in Primary Human Airway Epithelia. J Biol Chem [serie en Internet]. 2015 May 29 [citado 28 de Marzo de 2016];290(22). Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4447984/
22. Farinha CM, Sousa M, Canato S, Schmidt A, Uliyakina I, Amaral MD. Increased efficacy of VX-809 in different cellular systems results from an early stabilization effect of F508del-CFTR. Pharmacol Res Perspect [serie en Internet]. 2015 August [citado 28 de Marzo de 2016];3(4). Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4492728/
Recibido: 23 de mayo de 2016.
Aprobado: 8 de noviembre de 2016.
Elayne Esther Santana Hernández. Centro Provincial de Genética Médica. Avenida de Libertadores # 91, reparto Peralta, municipio Holguín. Holguín, Cuba. Correo electrónico: elsantana@infomed.sld.cu

