










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr vol.89 no.4 Ciudad de la Habana oct.-dic. 2017
PRESENTACIÓN DE CASO
Nefroblastoma o tumor de Wilms teratomatoso
Nephroblastoma or teratoid Wilms´tumor
Luis Alexis Graverán Sánchez,I Caridad Verdecia Cañizares,II Magda Alonso Pírez,II Damián Pineda FernándezIII
IServicio de Cirugía Pediátrica del Hospital Pediátrico Universitario "William Soler". La Habana, Cuba.
IIServicio de Oncocirugía del Hospital Pediátrico Universitario "William Soler". La Habana, Cuba.
IIIDepartamento de Anatomía Patológica del Hospital Pediátrico Universitario "William Soler". La Habana, Cuba.
RESUMEN
Introducción: el tumor de Wilms (nefroblastoma) es un tumor embrionario maligno de estirpe mesodérmica, que se origina en el riñón, probablemente, por una proliferación anormal del blastema metanéfrico. Es típico de la infancia, originado de remanentes renales inmaduros y compuestos por blastema renal, túbulos displásticos y soporte mesenquimal o estromal. Se manifiesta en pacientes en edad preescolar, edad media de 3,5 años, con una masa abdominal palpable asintomática y poco móvil, que no cruza línea media. Puede producir hematuria macroscópica e hipertensión arterial.
Presentación del caso: paciente femenina, mestiza, a la que se le diagnostica esta variante de tumor a muy temprana edad (solo 2 meses), descubierta por la madre y sin manifestaciones clínicas aparentes, como no fuera la gran distensión abdominal. Se logra establecer el diagnóstico mediante biopsia aspirativa con aguja fina, la cual plantea la existencia de tumor de Wilms teratomatoso, variante histológica poco frecuente, y quedó demostrada la intratabilidad con el empleo de agentes citorreductores probados en el tratamiento de esta neoplasia. Se opera vía laparotómica, y se reseca gran tumor que ocupaba toda la cavidad abdominal izquierda y cruzaba hacia la derecha, midió 28 x 15 x 10 cm de diámetro, y pesó 1 kg.
Conclusión: existen pocos casos publicados en la edad pediátrica con esta variante de neuroblastoma, y se resalta la importancia de la cirugía en la cura de la enfermedad.
Palabra clave: tumor de Wilms teratomatoso; Pediatría.
ABSTRACT
Introduction: Wilms´tumor (nephroblastoma) is a malignant embryonal tumor of mesodermal lineage, originated in the kidney probably because of abnormal proliferation of the metanephric blasthema. It is tipical of childhood, originating from immature renal remnants and composed by renal blasthema, displasic tubules and mesenchymal or stromal support. It manifests in patients at preschool age, average age of 3.5 years, with asymptomatic palpable abdominal mass of low mobility that does not cross the midline. It can cause macroscopic hematuria and blood hypertension.
Case presentation: female patient, mixed race who is diagnosed with this tumor variant at very small age (just two months); the disease was discovered by her mother, with no apparent clinical signs but great abdominal distension. It was possible to set the diagnosis through fine needle aspiration biopsy which revealed the existence of teratoid Wilms´tumor, an uncommon histological variant. It was proved that this disease could not be treated with the cytoreducing drugs used in the treatment of this neoplasia. The patient was operated on through laparotomy and a big rumor was resected, which occupied the whole left abdominal cavity and crossed into the right one; it measured 28 x 15 x 10 cm of diameter and weighed 1 kilogram.
Conclusions: there are few cases published at pediatric age with this type of neuroblastoma and the importance of surgery to cure the disease is stressed.
Keywords: teratoid Wilms´tumor; Pediatrics.
INTRODUCCIÓN
El tumor de Wilms o nefroblastoma es la neoplasia maligna renal más frecuente en niños. Su incidencia anual en Cuba es de aproximadamente 16 a 20 casos en niños menores de 15 años de edad, para una tasa de 0,7 por 100 000 habitantes.1 Es un tumor embrionario maligno, de estirpe mesodérmica, que se origina en el riñón, probablemente por una proliferación anormal del blastema metanéfrico, precursor del tejido renal normal definitivo, de carácter embrionario. Se origina por remanentes renales inmaduros, compuestos por blastema renal, túbulos displásticos y soporte mesenquimal o estroma. Existe una variante extremadamente rara en edad pediátrica que es el nefroblastoma o tumor de Wilms teratomatoso.
Los teratomas son neoplasias que se originan a partir de células pluripotenciales con diferentes líneas germinales embrionarias, comúnmente surgen en gónadas, región sacrococcígea, glándula pineal y retroperitoneo, entre otros sitios.2 Es muy raro su origen a partir de restos nefrogénicos.2,3
El nefroblastoma o tumor de Wilms en la infancia es un modelo de los tumores en el niño que responden bien al tratamiento oncoespecífico: quimioterapia y radioterapia. Actualmente el 90 % de los niños con nefroblastoma se curan, sin secuelas graves, gracias al empleo de la quimioterapia preoperatoria citorreductora, a las técnicas quirúrgicas cada vez más desarrolladas y a la reducción de las dosis de irradiación, sin embargo la variante teratomatosa no responde bien al tratamiento, por tener restos maduros (poco quimiosensibles) de diferentes hojas embrionarias del desarrollo fetal, por lo cual la cirugía es preponderante en su tratamiento.3-5
El diagnóstico de esta variedad de tumor es fácil. Ante un gran tumor abdominal localizado a uno de los flancos, de crecimiento rápido, que puede acompañarse de hematuria o no, en un niño que conserva buen estado general y que la madre fortuitamente durante el baño (o por el médico de asistencia durante un examen físico porque acude a la consulta generalmente por otro motivo), se hace el diagnóstico de un tumor abdominal. Los estudios imagenológicos modernos, sobre todo la ecografía y la tomografía axial computarizada (TAC), confirman el diagnóstico del nefroblastoma típico y del nefroblastoma teratomatoso, este último por la presencia de gran cantidad de elementos procedentes de tejidos en formación: hueso, cartílago, grasa, entre otros.6,7
Existen divergencias en relación con el empleo de la biopsia aspirativa con aguja fina (BAAF). Es controvertido, pero en nuestro medio no han ocurrido diseminaciones del tumor, y en manos de patólogos experimentados, el diagnóstico se ha establecido con un alto índice de certeza en pacientes con tumor de Wilms. Otros autores preconizan su uso en el diagnóstico de esta variante de tumor.8 El objetivo de este trabajo es presentar a la comunidad médica una paciente con esta variante rara de tumor de Wilms en etapa de lactante, y enfatizar en el rol de la cirugía como tratamiento definitivo de esta neoplasia.
PRESENTACIÓN CLÍNICA
Paciente de 2 meses de edad, sexo femenino y raza mestiza, con peso corporal en el momento del ingreso de 4,5 kg, que es traída de urgencia al cuerpo de guardia de nuestro hospital porque la madre le detectó distensión abdominal el 29 de noviembre de 2016 en horas de la noche. Fue evaluada por la guardia de Pediatría y Cirugía, quienes comprobaron gran tumor que ocupaba prácticamente todo el hemiabdomen izquierdo y cruzaba la línea media del abdomen. Se auscultaron los ruidos hidroaéreos hacia la derecha, que elevaron el diafragma y causaron cierta dificultad ventilatoria, por lo que se decide ingreso en unidad de cuidados intensivos polivalente (UTIP) (figura 1).

Se realiza ultrasonido de urgencia a su llegada, que informa: gran tumor heterogéneo, con áreas quísticas, tabiques, calcificaciones y elementos sólidos, que ocupaba todo el hemiabdomen izquierdo y cruza la línea media hacia la derecha. No se visualiza riñón izquierdo, y fue imposible realizar la mensuración de dicho tumor.
- Estudios de sangre: hemoglobina en 10,6g/L, cifras normales de plaquetas y leucocitos, hemoquímica dentro de parámetros normales.
- Determinación de marcadores tumorales: deshidrogenasa láctica, alfafetoproteína y fracción Beta de la gonadotrofina coriónica de valores normales.
- Rx de abdomen: asas desplazadas a la derecha, radiopacidad con calificaciones en proyección del hemiabdomen izquierdo.
- Rx de tórax: no alteraciones pleuropulmonares, solo acortamiento del diámetro torácico por elevación del diafragma.
- TAC de abdomen (30 de noviembre de 2016): imagen con densidades variables que ocupa todo el hemiabdomen izquierdo, se extiende desplazando estructuras vasculares y asas intestinales hacia la derecha, el riñón izquierdo se visualiza rechazado hacia la fosa iliaca ipilateral, y se observa que el tumor surge del mismo. Existen componentes quísticos dentro de la lesión con formación de tabiques y algunas calcificaciones dispersas.
Se realizan dos BAAF (1º y 7 de diciembre de 2016) bajo control de ultrasonido, que fueron positivas de células malignas, sin evidenciar variante citológica. Se discutió en equipo multidisciplinario, y dado el gran volumen tumoral y la irresecabilidad del tumor, se inicia tratamiento citorreductor para un tumor embrionario a un tercio de la dosis de ciclofosfamida (CFM), vincristina (VCR) y actinomicin D (ACTD), pero solo se logra una mínima reducción. La paciente en la UTIP requirió trasfusión de glóbulos en dos ocasiones durante la quimioterapia por cifras bajas de hemoglobina.
Se intenta una nueva BAAF el 12 de diciembre de 2016, que evidenció tumor de Wilms teratomatoso, y entonces se rediscute y se decidió administrar el tratamiento preoperatorio para el nefroblastoma estadio avanzado, con bajas dosis semanales de VCR y ACTD, y su traslado al servicio de Oncocirugía. Se fue evaluando su respuesta semanalmente durante 4 semanas, sin reducción apreciable del diámetro tumoral ni de la circunferencia abdominal, por el contrario, de 44 cm iniciales aumentó a 50, por lo que se discute nuevamente en el seno del comité de tumores, y se decide, por todo el equipo médico que lo integra, ante la progresión tumoral, llevar al salón de operaciones para obtener tejido para estudio histológico definitivo, con la resección del tumor y/o biopsia en caso de que fuera irresecable.
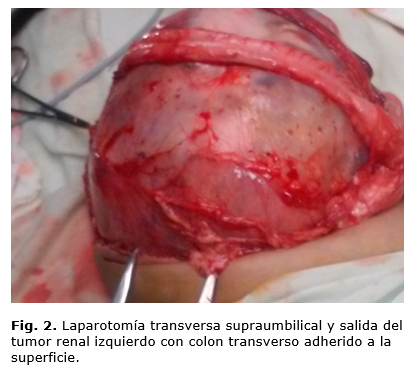
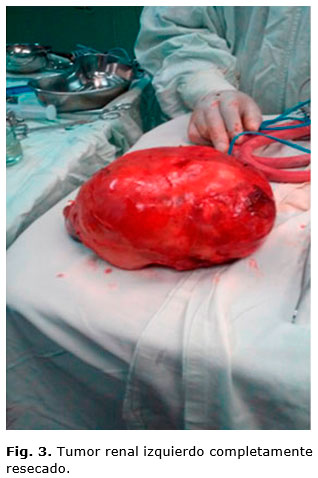
Se opera el 12 de enero de 2017. Se realiza laparotomía amplia transversa supraumbilical, y se encuentra gran tumor rojo carnoso, con superficie algo irregular y bolsones quísticos en su interior, áreas blanquecinas duras, de un diámetro mayor de 28 x 15 x 10 cm, que ocupaba hipocondrio, flanco y fosa iliaca izquierda, cruzando la línea media y comprimiendo asas intestinales delgadas hacia la derecha, con aorta abdominal y cava inferior también desplazadas hacia el mismo lado. No había ganglios ni líquido libre en la cavidad, tenía el colon transverso adherido a su superficie superior, pero se logra separar cuidando bien su vascularización (figura 2). El tumor fue resecado completamente, liberándolo de adherencias establecidas con estructuras cercanas, y ligando uréter y vasos del hilio renal izquierdo (figura 3). El peso del tumor fuera de la cavidad abdominal fue de 1 kg.
La paciente fue llevada en el posoperatorio inmediato a la UTIP, sin requerir ventilación, mantuvo diuresis adecuada, y se le administró antibioticoterapia con ceftriaxona vía endovenosa. Presentó al tercer día de operada una enteritis grave que requirió apoyo inotrópico y cambio a otros antibióticos (vancomicina y meropenen), sonda nasogástrica y reporte de crítico durante varios días. Después las deposiciones fueron siendo moldeadas, y se inició la alimentación vía enteral hasta que sale de la UTIP el 26 de enero para el servicio de Oncología con mejoría clínica evidente.
El resultado de Anatomía Patológica fue: nefroblastoma o tumor de Wilms teratomatoso, que contenía elementos de las tres hojas embrionarias (tejido encefálico inmaduro [elementos gliales], elementos de tejido digestivo, piel, cartílago, uñas, fragmentos de huesos, focos de cartílago, entre tejido renal inmaduro), que midió 28 x 15 x 10 cm.
DISCUSIÓN
El nefroblastoma puede ser de histología favorable o desfavorable, de ahí su evolución y respuesta al tratamiento. La variante teratomatosa es extremadamente rara en edad pediátrica, pero generalmente de histología favorable, por contener elementos de diversos tejidos maduros. Karima Idrissi y otros3 publicaron un caso muy similar al nuestro, un teratoma renal primario como una rara entidad, detectada en un niño de 6 meses de edad, que se atendía por dolor intermitente y distensión abdominal desde el primer mes de nacido. Al examen físico tenía masa palpable en flanco e hipocondrio izquierdo, de bordes irregulares. Le realizaron estudios de hemoquímica dentro de límites normales, radiografía de tórax normal, y en el ultrasonido abdominal se detectó tumor renal izquierdo de 18 cm de diámetro, con cavidades quísticas y componentes sólidos en su interior.
Al no encontrarse otras alteraciones en el resto de los órganos intraabdominales, y pensando en que a esta edad es el nefrona mesoblástico el tumor renal más frecuente o un tumor de Wilms, se decide operarlo. Se le realizó laparotomía supraumbilical, y se observó gran tumor sólido y quístico con áreas de hemorragias proveniente de la pelvis renal izquierda, fue resecado y el resultado anomopatológico fue tumor de 18 x 12 x 8 cm de diámetro con peso de 200 g, y al corte, con áreas quísticas en su interior y gran cantidad de elementos sólidos. El estudio microscópico mostró la presencia de tejido escamoso quertinizado, focos cartílago, tejido epitelial columnar mucinoso, formación de hueso, melanina, focos de tejido inmaduro neuroectodérmico y áreas quísticas con tejido renal inmaduro en su interior, por lo que se diagnostica teratoma renal.
La mayoría de los autores plantean3-5 que esta variante de tumor puede detectarse antes del nacimiento por estudios prenatales, que ocasionalmente pueden observarse después del nacimiento por manifestaciones clínicas, como: dolor abdominal, disuria, hematuria y constipación; o pueden diagnosticarse en el curso de un examen físico de abdomen fortuitamente, cuando el tumor ha alcanzado cierto diámetro, pero se confirman mediante la radiografía de abdomen, en la que se visualiza calcificación en proyección renal, o la presencia de la masa tumoral detectada mediante ultrasonido.
Los teratomas se originan generalmente en el retroperitoneo, región sacrococcígea y mediastino, pero raros a nivel del riñón.2,4,6,7 Los estudios topográficos permiten una mejor visualización del tumor y de las estructuras que lo componen.8-10 Algunos autores plantean que el empleo de la angiografía y cavografía, así como la BAAF, son necesarios para realizar el diagnóstico de esta entidad.8,11 La determinación en suero de marcadores tumorales como la alfafetoproteína, que suele estar elevada en estos pacientes y una vez resecado el tumor disminuye, es un índice para detectar su recurrencia.12
Hay que establecer el diagnóstico diferencial entre el teratoma dentro de un tumor de Wilms y el nefroma mesoblástico mediante examen histológico. El nefroblastoma típico debe tener elementos blastemales, epitelio y elementos estromales, mientras que entre el teratoma intrarrenal y el nefroblastoma teratoide solo se logra la diferenciación mediante el estudio anatomopatológico del tumor, después de la resección, ya que contienen diversos elementos de tejido procedente de las tres hojas embrionarias del desarrollo fetal intrauterino. El nefroma mesoblástico tiene una apariencia ultrasonográfica típica de tumor no encapsulado, con excelente pronóstico, se observa generalmente en pacientes de menores de 3 meses de edad, y se cura con la cirugía. También hay que diferenciarlo del neuroblastoma quístico, del adenoma metanéfrico, de un linfoma renal, de un rabdomiosarcoma, de un sarcoma de células claras y de un carcinoma renal primario, estos últimos de un pobre pronóstico.3,4
Beckwith5 planteó que el término teratoma renal es un tumor de origen intrarrenal, con cápsula y elementos de las tres hojas embrionarias del desarrollo fetal, que resuelven con la completa resección quirúrgica, y no es necesario -en la mayoría de los casos- la administración de tratamiento quimioterápico, con una supervivencia elevada después de la cirugía completa del tumor de 85 %.5-7
Existe un reporte de caso de un paciente con tumor de Wilms bilateral, en el cual el tumor renal derecho tenía el clásico patrón trifásico del nefroblastoma, pero se detectó en el tumor del riñón izquierdo un foco de elementos de teratoma renal, demostrado mediante biopsia percutánea.8 También se han reportado formas puramente quísticas del tumor renal teratoide con una evolución favorable.9,10
Biswanath y otros11 reportaron un caso muy similar al nuestro en un paciente de 4 años de edad, pero de localización renal derecha, que no cruzó la línea media del abdomen, con un diámetro de 10 cm, mucho menor al diámetro reportado por nosotros. Fernández y otros4 definieron el tumor de Wilms teratoide como un tumor heterólogo compuesto de: tejido adiposo, tejido glial, músculo, con 50 % de cartílago o hueso dentro de la neoplasia renal.
Nuestro caso tenía múltiples factores pronósticos en contra. Primero, la temprana edad de presentación (con solo dos meses de edad), que hizo que no se pudiera administrar el tratamiento a la dosis necesaria para lograr la reducción tumoral, debido a la inmadurez hepática y renal propia de la lactancia, el estadio avanzado de la enfermedad con que nos llega la paciente (estadio III), abdomen extenso con compresión del intestino, y desplazamiento de estructuras vitales como aorta y vana cava inferior, adherido al colon transverso y con compresión del bazo, pero afortunadamente se logró la resección del gran tumor y se liberó de todos los elementos antes mencionados (el bazo mejoró su color al quitar la compresión a que estaba sometido). Además, la variante citológica es poco frecuente a esta edad.
Los niveles de alfafetoproteína en nuestra paciente estaban dentro de los límites normales, pero muchos autores publican que generalmente están elevados por su origen, lo cual sirve, una vez tratado el caso, para el seguimiento de recurrencia.12 Existen publicaciones en las que mencionan que la quimioterapia puede ser administrada en casos de tumor de Wilms teratomatoso dependiendo del tamaño del tumor, estadio clínico, edad del paciente al diagnóstico y la histología, pero generalmente es baja su respuesta a los agentes citostáticos y radiaciones.13-15
El tratamiento del tumor de Wilms teratoide no ha sido bien establecido, porque es una entidad rara y por la presencia de varios componentes maduros heterólogos, que lo hacen resistente a la quimioterapia y a la radioterapia, por lo cual la cirugía es el tratamiento más utilizado.16-18
Se concluye que, a pesar de su rareza, el nefroblastoma teratomatoso puede presentarse a muy temprana edad de la vida, y que su respuesta al tratamiento con quimioterapia puede ser pobre o nula, debido a los componentes de diversos tejidos en formación del organismo, por lo que la cirugía es el rol curativo en estos casos.
Conflicto de intereses
Los autores declaran no tener conflicto de intereses en la realización del estudio.
REFERENCIAS BIBLIOGRÁFICAS
1. Cuba. Ministerio de Salud Pública (Minsap). Anuario estadístico de salud, 2015 [homepage en Internet]; Dirección Nacional de Registros Médicos [citado 15 de febrero de 2017]. Disponible en: https://www.researchgate.net/publication/301226389_Anuario_estadistico_de_Salud_2015
2. Jones NM, Kiely EM. Retroperitoneal teratomas-potential for surgical misadventure. J Pediatr Surg. 2008;43:184-6.
3. Idrissi-Serhrouchni K, El-Fatemi H, El Madi A, Chbani L, Harmouch T. Primary renal teratoma: a rare entity. Diagnostic Pathology. 2013;8:107.
5. Beckwith JB. Wilms' tumor and other renal tumors of childhood: A selective review from the National Wilms' tumor Study Pathology Center. Hum Pathol. 1983;14:481-92.
6. Mandal KC, Mukhopadhyay M, Barman S, Halder P, Mukhopadhyay B, Kumar R. Uncommon renal tumors in children: A single center experience. J Indian Assoc Pediatr Surg. 2016;21:61-5.
7. Amit C, Samir M, Ashish W, Tandon RK, Wakhlu AK. Retroperitoneal Teratomas in Children. Indian J Pediatr. 2006;73:221-3.
8. Wang H, Li F, Liu J, Zhang S. Ultrasound-guided core needle biopsy in diagnosis of abdominal and pelvic neoplasm in pediatric patients. Pediatr Surg Int. 2014;30:31-7.
9. Emerson RE, Kao CS, Eble JN, Grignon DJ, Wang M, Zhang S, et al. Evidence of a dual histogenetic pathway of sacrococcygeal teratomas. Histopathology. 2017;70:290-300.
10. Richards ML, Gundersen AE, Williams MS. Cystic neuroblastoma in infancy. J Pediatr Surg. 1995;30:1354-7.
11. Biswanath M, Ram MS, Madhumita M, Sabitri M, Roy D, Malay KB. Teratoid Wilms' tumor-A rare renal tumour.Urol Ann. 2011;3:155-7.
12. Parikh B, Trivedi P, Shukla K. A unilateral teratoid Wilms' tumor with raised serum alpha-fetoprotein level. Indian J Pathol Microbiol. 2007;50:317-9.
13. Gupta R, Sharma A, Arora R, Dinda AK. Stroma-predominant Wilms tumor with teratoid features: Report of a rare case and review of the literature. Pediatr Surg Int. 2009;25:293-5.
14. Seo J, Suh YL, Choi HY. Adult teratoid Wilms' tumor with prominent neuroepithelial differentiation. Pathol Int. 2009;59:44-8.
15. Inoue M, Uchida K, Kohei O, Nashida Y, Deguchi T, Komada Y, et al. TeratoidWilms' and apos: A case report with literature review. J Pediatr Surg. 2006;41:1759-63.
16. Karakuş E, Senayli A, Özcan F, Demir AH, Tiryaki T, Özyörük D, et al. Teratoid Wilms' tumor exhibiting extensive squamous differentiation.Fetal Pediatr Pathol. 2015;34:70-2.17. Yadav YK, Sharma U, Gupta K, Arora R. Squamous predominant teratoid Wilms' tumor. J Lab Physicians. 2012;4:50-2.
18. Ishida M, Hotta M, Ohta M, Taga T, Ohta S, Takeuchi Y, et al. A case of retroperitoneal immature teratoma with nephroblastic components. J Pediatr Hematol Oncol. 2012;34:22-5.
Recibido: 27 de febrero de 2017.
Aprobado: 31 de marzo de 2017.
Luis Alexis Graverán Sánchez. Hospital Pediátrico Universitario "William Soler". San Francisco # 10 112, reparto Altahabana, municipio Boyeros. La Habana, Cuba. Correo electrónico: alexisgs@infomed.sld.cu

